Nel caso dell'interazione di due atomi:
U è l'energia di interazione;
U = U + TIRO.
 - Equazione di Lennard-Jones
, c, b, m = cost
- Equazione di Lennard-Jones
, c, b, m = cost
Nei casi di interazione di atomi con una superficie solida, è necessario riassumere tutte le interazioni.
x è la distanza dalla superficie
r - il raggio d'azione delle forze di attrazione
dV - volume
n è il numero di molecole di superficie
U ADS. è l'energia di interazione di adsorbimento



Nel caso dell'adsorbimento, l'attrazione aumenta. E nel caso di interazione di tipo non polare-non polare, l'adsorbimento è prevalentemente localizzato nelle depressioni.
interazione elettrostatica.
Adsorbente polare - adsorbato non polare
Adsorbente non polare - adsorbato polare
Un adsorbente polare è un adsorbato polare.
M  la molecola di adsorbato è rappresentata come un dipolo, e l'adsorbente come un conduttore, in cui la molecola di adsorbato induce un dipolo simmetrico rispetto a quello dato.
la molecola di adsorbato è rappresentata come un dipolo, e l'adsorbente come un conduttore, in cui la molecola di adsorbato induce un dipolo simmetrico rispetto a quello dato.
X - distanza dal centro
Quando si interagisce, sorge il potenziale:
 ,
,
 è il momento di dipolo.
è il momento di dipolo.
Il potenziale tende ad assumere il valore massimo, cioè i dipoli tendono ad orientarsi perpendicolarmente alla superficie.
Poiché un aumento della temperatura favorisce la crescita del moto browniano, porta a una decelerazione del processo di adsorbimento.
Nel caso di interazione elettrostatica, l'adsorbato è localizzato prevalentemente su sporgenze.
Equazione fondamentale di adsorbimento.
In caso di adsorbimento, il componente viene ridistribuito, il che significa che il potenziale chimico cambia. Il processo di adsorbimento può essere considerato come la transizione dell'energia superficiale in energia chimica.
Volume strato = 0, quindi l'equazione generalizzata I e II della legge della termodinamica:
T = cost; (1) = (2) => 

Per un sistema a due componenti:
,
 ,
,
 =>
=>
=>
 - Equazione di adsorbimento di Gibbs
.
- Equazione di adsorbimento di Gibbs
.
Per il caso di adsorbimento della TV. corpo - gas:, 
 ,
,

 - isoterma
- isoterma
 - isobare
- isobare
 - isopicno
- isopicno
 - isostere
- isostere
Isotherm, isopycne, isostere sono correlati tra loro.
Perché funzione di adsorbimento 



Isoterma di Henry Isoterma di Langmuir



Termodinamica. Adsorbimento.
Per mezzi condensati:
 ,
,
 ,
,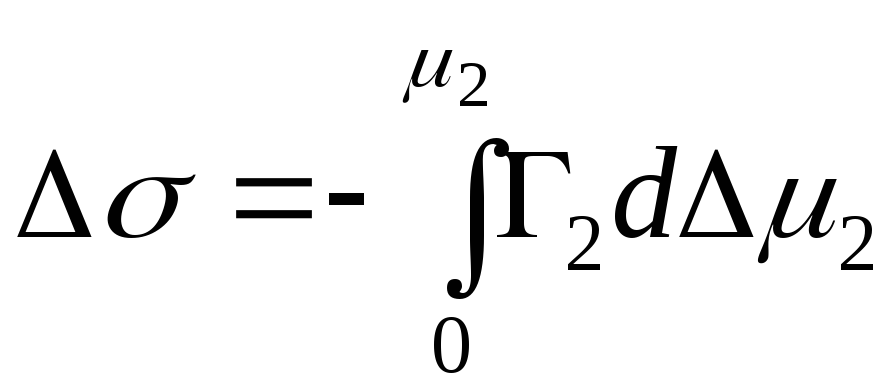
 - cambiamento integrale nell'energia di Gibbs
.
- cambiamento integrale nell'energia di Gibbs
.

P-pressione su una superficie curva, P S-pressione su una superficie piana
 - potenziale di adsorbimento
- potenziale di adsorbimento
Cambio differenziale in intrappolamento 
 , Ã = cost
, Ã = cost
- variazione di entropia differenziale
- entalpia differenziale di adsorbimento
 - calore isosterico di adsorbimento
- calore isosterico di adsorbimento
 - calore di condensazione
- calore di condensazione
 - calore netto di adsorbimento
- calore netto di adsorbimento

 ,
,




Qa è il calore integrale di adsorbimento, 
Qra è il calore netto integrale di adsorbimento,

L'equazione di Henry
Lo studio dell'adsorbimento è ostacolato dalla disomogeneità della superficie, quindi si ottengono le regolarità più semplici per superfici omogenee.
Consideriamo l'interazione dei gas con una superficie solida, quando un gas passa da uno stato di equilibrio nel volume ad uno stato di equilibrio sulla superficie. Questo caso è analogo all'equilibrio dei gas in un campo gravitazionale.
 ,
,
 ,
=>
,
=> -L'equazione di Henry
-L'equazione di Henry
 - coefficiente di distribuzione
- coefficiente di distribuzione
Nel processo di adsorbimento, si verifica un cambiamento nei potenziali chimici.
Per la fase di massa: 
Per gas di superficie: 
In uno stato di equilibrio  , cioè.
, cioè.

Nell'equazione di Henry, la costante non dipende dalla concentrazione
L'equazione di Henry è valida nella regione delle basse pressioni e concentrazioni. All'aumentare della concentrazione, sono possibili 2 tipi di deviazioni dalla legge di Henry:
1 - deviazioni positive, D diminuisce, A diminuisce
2 - deviazioni negative, D - aumenta, A - aumenta.
Il tipo di deviazione è determinato dalla predominanza dell'uno o dell'altro tipo di interazione adsorbente-adsorbato.
Con una forte interazione adesiva, i coefficienti di attività aumentano - una deviazione positiva. Nel caso di interazioni coese, si osservano deviazioni negative.
adsorbimento monomolecolare.
Isoterma di Langmuir.
Le regolarità più semplici sono state ottenute nella teoria di Henry. Langmuir ha proposto una teoria secondo la quale l'adsorbimento è considerato una reazione quasi chimica. in cui:
La superficie è energeticamente uniforme.
L'adsorbimento è localizzato, ogni centro di adsorbimento interagisce con una molecola di adsorbato.
Le molecole di adsorbato non interagiscono tra loro.
L'adsorbimento è monostrato.

 - superficie,
- superficie,  - adsorbato,
- adsorbato,  - complesso di adsorbimento.
- complesso di adsorbimento.
 , quindi la concentrazione dei siti di adsorbimento:
, quindi la concentrazione dei siti di adsorbimento:  ,
, - limitare l'assorbimento.
- limitare l'assorbimento.
 , quindi la costante di reazione:
, quindi la costante di reazione: 

 - Equazione di Langmuir.
- Equazione di Langmuir.
Adsorbimento contro concentrazione
1 )
)
 ,
,
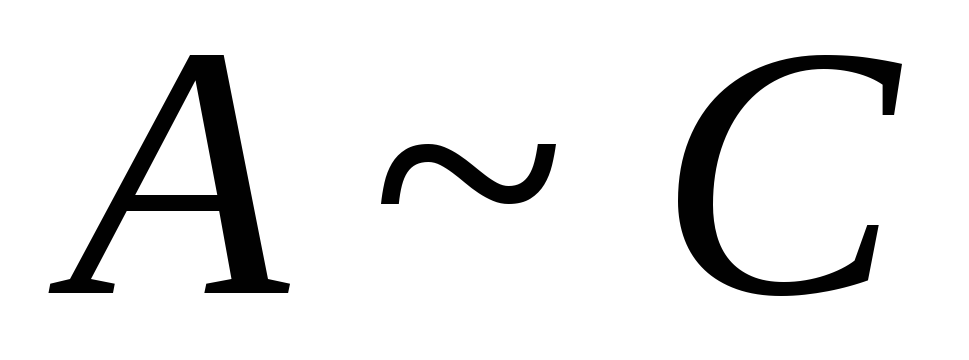
2) area di alte concentrazioni
 - adsorbimento limitante, formazione di uno strato monomolecolare
- adsorbimento limitante, formazione di uno strato monomolecolare
Per l'energia di Gibbs: .
g è il fattore di entropia.
Nel caso dell'isoterma di Henry, l'energia di Gibbs caratterizza la transizione dell'adsorbato dallo stato standard nella massa allo stato standard sulla superficie. Nel caso dell'isoterma di Langmuir  caratterizza il grado di affinità dell'adsorbente e dell'adsorbato.
caratterizza il grado di affinità dell'adsorbente e dell'adsorbato.
 trovato dall'isobar di van't Hoff.
trovato dall'isobar di van't Hoff.

 , poi
, poi  , quindi
, quindi  .
.

 - grado di riempimento superficiale.
- grado di riempimento superficiale.




 - il numero di posti vacanti,
- il numero di posti vacanti,  - il numero dei posti occupati.
- il numero dei posti occupati.
 ,
,

Quelli. nella regione di alte concentrazioni, il numero di siti liberi è inversamente proporzionale alla quantità di adsorbato.
Adsorbimento di una miscela di gas su una superficie omogenea.
In questo caso, il processo di adsorbimento è considerato come due reazioni parallele.
 (1)
(1)

 (2)
(2)


Adsorbimento di una miscela di gas su una superficie disomogenea.
Nel caso di una superficie non omogenea non ci si deve limitare a tamponamenti medi.
Come risultato della concorrenza, la localizzazione di vari adsorbati è possibile in diversi tipi di siti.
In questo caso, la relazione  .
.
 ,
,
 è la pressione di vapore di saturazione dell'adsorbato.
è la pressione di vapore di saturazione dell'adsorbato.
 ,
,
 è il calore di adsorbimento.
è il calore di adsorbimento.
"+" - dipendenza simbatica, "-" - dipendenza antibatica, "H" - nessuna correlazione.
"+" - l'adsorbimento procede secondo lo stesso meccanismo. Nelle zone energeticamente più favorevoli viene adsorbito prevalentemente un gas con elevata affinità alla superficie.
"-" - l'adsorbimento avviene attraverso vari meccanismi e fino a un certo momento non c'è competizione per la superficie.
L'adsorbimento monomolecolare si realizza prevalentemente durante l'adsorbimento fisico di gas a bassi valori p, nonché all'interfaccia liquido/gas.
Adsorbimento polimolecolare.
Teoria della SCOMMESSA(Brunauer, Emmet, Cassiere).
Nel caso in cui la formazione di un monostrato non sia sufficiente a compensare l'energia superficiale, l'adsorbimento è polimolecolare e può essere considerato come il risultato di una condensazione forzata sotto l'azione delle forze superficiali.
Disposizioni di base:
Quando una molecola di adsorbato colpisce il sito occupato, si forma un insieme multiplo.
Man mano che ti avvicini p a p S il numero di siti di adsorbimento liberi diminuisce. Inizialmente il numero dei posti occupati da singoli, doppi, ecc. aumenta e poi diminuisce. kit.
In p =p S l'adsorbimento si trasforma in condensazione.
Non ci sono interazioni orizzontali.
Per il primo strato viene eseguita l'isoterma di Langmuir.
La superficie è considerata come un insieme di siti di adsorbimento. La condizione di equilibrio dinamico è valida: la velocità di condensazione nei luoghi liberi è uguale alla velocità di evaporazione da quelle occupate.
a è il coefficiente di condensazione (la frazione di molecole condensate sulla superficie);
 ,
,
Zm è il numero massimo di posti liberi.
 - frequenza delle vibrazioni degli atomi nella direzione perpendicolare alla superficie.
- frequenza delle vibrazioni degli atomi nella direzione perpendicolare alla superficie.
Per il primo strato, le condizioni di equilibrio dinamico sono:



 , poi
, poi
 - Equazione di Langmuir.
- Equazione di Langmuir.
Per il secondo strato sarà vero: 
Per l'i-esimo strato: 
Per semplicità si assume che a e ν siano gli stessi per tutti gli strati tranne il primo. Per tutti gli strati tranne il primo, il calore di adsorbimento è costante. Per l'ultimo strato, il calore di adsorbimento è uguale al calore di condensazione. Di conseguenza, l'equazione
 (*)
(*)
C- costante,
Nel caso della teoria BET, la costante Insieme a caratterizza l'energia di Gibbs di puro adsorbimento. L'equazione contiene solo una costante e questa equazione è anche molto importante per determinare la superficie specifica dell'adsorbente.
Poiché il calore viene rilasciato per adsorbimento, la determinazione delle superfici specifiche viene effettuata a basse temperature.
 ????????????
????????????


Il difetto principale della teoria– Trascurare le interazioni orizzontali a favore di quelle verticali.
L'equazione è nell'intervallo  da 0,05 a 0,3.
da 0,05 a 0,3.
In cui si  <
0,05 – существенное влияние оказывает
неоднородность поверхности.
<
0,05 – существенное влияние оказывает
неоднородность поверхности.
 > 0,3 - l'interazione dell'adsorbato - influenza l'adsorbato.
> 0,3 - l'interazione dell'adsorbato - influenza l'adsorbato.
Contabilità delle interazioni adsorbato-adsorbato.
Le interazioni compaiono durante l'adsorbimento su una superficie non polare di molecole o molecole ramificate. capace di formare associazioni. In questo caso, la forma delle isoterme di adsorbimento cambia.
MA  il assorbente non è polare.
il assorbente non è polare.
Il grafico 1 corrisponde alle interazioni deboli adsorbato-adsorbato, forte adsorbato-adsorbente.
Il grafico 2 corrisponde a una forte interazione adsorbato-adsorbato, una forte interazione adsorbato-adsorbente.
Il grafico 3 corrisponde a una forte interazione adsorbato-adsorbato, una debole interazione adsorbato-adsorbente.
 ,
,

Nel caso di interazione tra molecole di adsorbato, è necessario tenere conto delle variazioni dei coefficienti di attività. E questa equazione è scritta come:
 - l'equazione di Frunkin, Fowler, Guggenheim.
- l'equazione di Frunkin, Fowler, Guggenheim.
Kè la costante di attrazione.
La teoria del potenziale di Polan.
Questa teoria non fa derivare alcun tipo di isoterma di adsorbimento, ma permette di calcolare isoterme a diversa temperatura.
Adsorbimentoè il risultato dell'attrazione dell'adsorbato sulla superficie dell'adsorbente per azione del potenziale di adsorbimento, che non dipende dalla presenza di altre molecole e dipende dalla distanza tra la superficie e la molecola di adsorbato.
 ,
,
 - potenziale di adsorbimento.
- potenziale di adsorbimento.
Poiché la superficie è disomogenea, la distanza viene sostituita dal volume di adsorbimento  .volume di adsorbimentoè il volume racchiuso tra la superficie e il punto corrispondente al valore dato
.volume di adsorbimentoè il volume racchiuso tra la superficie e il punto corrispondente al valore dato  .
.
Potenziale di adsorbimentoè il lavoro di trasferire 1 mole di adsorbato al di fuori del dato volume di adsorbimento in un dato punto del volume di adsorbimento (o il lavoro di trasferire 1 mole di vapore saturo dell'adsorbato, che è in equilibrio con l'adsorbato liquido in assenza di l'adsorbente, nella fase vapore in equilibrio con l'adsorbente).

Curva caratteristica
 - potenziale di adsorbimento,
- potenziale di adsorbimento, 
Per un dato adsorbente e vari adsorbati vale quanto segue: 
Per diversi tipi di adsorbati  ,
,
dove  potenziali di isoterme di adsorbimento a pressioni relative
potenziali di isoterme di adsorbimento a pressioni relative  per l'adsorbato 1 e per l'adsorbato 2. Questo rapporto è un valore costante.
per l'adsorbato 1 e per l'adsorbato 2. Questo rapporto è un valore costante.
 - coefficiente di affinità
- coefficiente di affinità
Teoria della condensazione capillare.
Il corso del processo di adsorbimento dipende in gran parte dalla struttura del corpo poroso.
|
microporoso | |
|
Poroso di transizione | |
|
Macroporoso |
Nel caso di assorbenti microporosi, i campi delle forze di adsorbimento si sovrappongono. Nel caso di assorbenti macroporosi, i pori fungono da canali di trasporto. I processi di condensazione sono più significativi nei corpi porosi transitori. La condensazione capillare inizia a determinati valori p e  quando parte dell'energia superficiale è già stata compensata. Condizione necessaria è che la superficie sia autoportante. Il processo è descritto Equazione Thompson-Kelvin.
quando parte dell'energia superficiale è già stata compensata. Condizione necessaria è che la superficie sia autoportante. Il processo è descritto Equazione Thompson-Kelvin.

 - nel caso di bagnatura, il centro di curvatura è in fase gassosa.
- nel caso di bagnatura, il centro di curvatura è in fase gassosa.
Nel caso di condensazione capillare, l'isoterma di adsorbimento ha una forma di isteresi. Il ramo inferiore corrisponde al processo di adsorbimento e il ramo superiore corrisponde al processo di desorbimento.
Tutti i tipi di pori possono essere ridotti a tre tipi:
|
conico |
Cilindrico con un'estremità chiusa |
Cilindrico con due estremità aperte |
|
Il riempimento del processo viene effettuato dal fondo del poro. L'isoterma di adsorbimento e l'isoterma di desorbimento in questo caso coincidono, poiché il processo di adsorbimento inizia con una sfera e anche il processo di desorbimento inizia con la scomparsa di alcune sfere.
↓ |
Non c'è isteresi. La corsa in avanti e indietro sono descritte dall'equazione:
|
Non c'è fondo da nessuna parte, il riempimento del poro andrà lungo le pareti del cilindro.
cilindro: isoterma e avrà una forma di isteresi.
↓ |


 - sfera,
- sfera, ,
,



A  condizioni di bagnatura, la condensazione avviene a pressioni inferiori, il che è energeticamente favorevole. Dal ramo di desorbimento si ottengono le curve di distribuzione della dimensione dei pori.
condizioni di bagnatura, la condensazione avviene a pressioni inferiori, il che è energeticamente favorevole. Dal ramo di desorbimento si ottengono le curve di distribuzione della dimensione dei pori.
Il massimo della curva differenziale viene spostato a sinistra rispetto al punto di flesso dell'integrale. Il volume totale dei piccoli pori è piccolo, ma ha grandi superfici. All'aumentare della dimensione dei pori, il loro volume aumenta come  , e l'area come
, e l'area come  , per questo motivo si osserva uno spostamento del massimo della curva differenziale.
, per questo motivo si osserva uno spostamento del massimo della curva differenziale.
Adsorbimento all'interfaccia solido-liquido.
Nel caso dell'adsorbimento all'interfaccia solido-gas, abbiamo trascurato un componente. Nel caso di adsorbimento all'interfaccia solido-liquido, l'adsorbato sposta le molecole di solvente dalla superficie dell'adsorbente.
 ,
,
L'equazione giusta è:

 ,
,
N 1, N 2 - frazioni molari del solvente e del componente, N 1 + N 2 \u003d 1, quindi
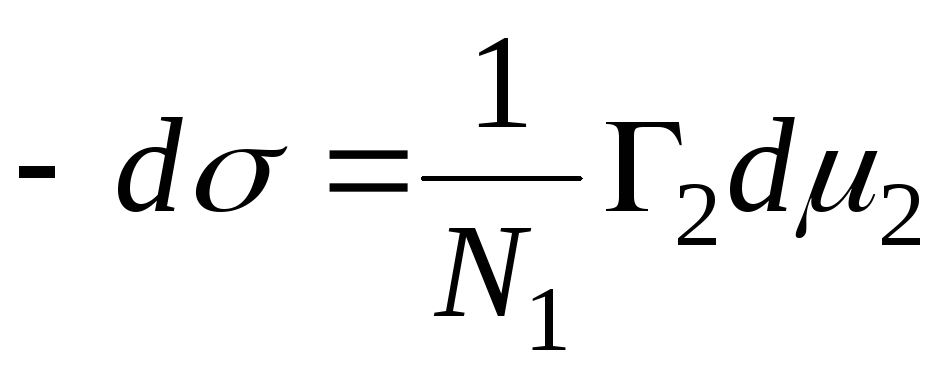 ,
=>
,
=>
 , quindi - l'equazione di adsorbimento per il confine di fase solido - liquido.
, quindi - l'equazione di adsorbimento per il confine di fase solido - liquido.
Adsorbimento (G) > 0 a  <
0
<
0
Se i valori  per il componente e il solvente sono molto diversi, in questo caso la dipendenza G a partire dal N ha un estremo al valore N
~ 0,5.
per il componente e il solvente sono molto diversi, in questo caso la dipendenza G a partire dal N ha un estremo al valore N
~ 0,5.
e  Se
Se  hanno valori simili, in questo caso il segno di adsorbimento può cambiare. Dipendenza G a partire dal N attraversa l'asse x
hanno valori simili, in questo caso il segno di adsorbimento può cambiare. Dipendenza G a partire dal N attraversa l'asse x
Intersezione di funzioni G(N) con l'asse delle ascisse viene chiamato adsorbimento azeotropico. Ciò significa che i due componenti non possono essere separati su questo adsorbente.
Equazione dell'isoterma di adsorbimento con costante di scambio.
Durante l'adsorbimento all'interfaccia solido-liquido, i componenti vengono costantemente ridistribuiti tra la superficie dell'adsorbente e il volume della soluzione.
 - componenti (- - riferiti alla superficie)
- componenti (- - riferiti alla superficie)

 ,
,
 ,
, .
.
 ,
,


Adsorbimento all'interfaccia liquido-gas
R  Consideriamo la variazione del profilo di concentrazione al passaggio dell'interfaccia liquido-gas. Lascia che il componente 2 sia volatile.
Consideriamo la variazione del profilo di concentrazione al passaggio dell'interfaccia liquido-gas. Lascia che il componente 2 sia volatile.
Cs è la concentrazione nello strato superficiale.
Basato sulla definizione di eccesso di adsorbimento

Se il componente non è volatile, il valore di adsorbimento sarà scritto come segue:
P  ri
ri 

Nell'equazione  la natura della materia è descritta dalla derivata
la natura della materia è descritta dalla derivata  .
.
L'isoterma di tensione superficiale può essere della forma 1 o 2:
1 - tensioattivi
2 - tensioattivi
L'attività superficiale g è la capacità delle sostanze di ridurre la tensione superficiale nel sistema.

 - spessore dello strato superficiale
- spessore dello strato superficiale
C Sè la concentrazione del componente nello strato superficiale
Insieme a– concentrazione in volume
Per una serie omologa c'è una regola:
 - Regola di Traubeau Duclos
- Regola di Traubeau Duclos
Per la serie omologa, l'isoterma di adsorbimento si presenta così:
Scriviamo D invece di A, poiché l'adsorbimento è eccessivo nello strato superficiale.
Isoterma di tensione superficiale:

 è la tensione superficiale del solvente puro.
è la tensione superficiale del solvente puro.
 - equazione fondamentale di adsorbimento;
- equazione fondamentale di adsorbimento;
 - Equazione di Langmuir.
- Equazione di Langmuir.
Risolviamoli insieme:

- L'equazione di Shishkovsky.
Bè una costante per la serie omologa.
UN- quando si passa da un omologo all'altro, aumenta di 3-3,5 volte 
![]()
1 - area di basse concentrazioni
![]()
2 - concentrazione media
3 - strato monomolecolare
I tensioattivi sono molecole anfifiliche, cioè includono un gruppo polare e un radicale idrocarburico non polare.
o è la parte polare della molecola.
| è la parte non polare della molecola.
In un solvente polare, le molecole di tensioattivo sono orientate in modo tale che la parte polare della molecola sia rivolta verso il solvente, mentre la parte non polare viene spinta nella fase gassosa.
Nell'equazione di Shishkovsky  , è costante per la serie omologa.
, è costante per la serie omologa.
L'azione tensioattiva inizia ad apparire con n>5. A concentrazioni superiori alla concentrazione dello strato monomolecolare, la micellizzazione avviene in soluzioni di tensioattivi.
Micella- viene chiamato l'aggregato di molecole di tensioattivo anfifilico, i cui radicali idrocarburici formano il nucleo e i gruppi polari vengono trasformati nella fase acquosa.
Massa micellare - massa micellare.
H  il numero di molecole è il numero di aggregazione.
il numero di molecole è il numero di aggregazione.
Micelle sferiche
Nel caso della micellizzazione, si stabilisce un equilibrio nella soluzione
CMC è la concentrazione micellare critica.
Poiché consideriamo le micelle una fase separata:
Per la serie omologica, esiste un'equazione empirica:
unè l'energia di dissoluzione del gruppo funzionale.
b è l'incremento del potenziale di adsorbimento, il lavoro di adsorbimento per unità di metilene.
è l'incremento del potenziale di adsorbimento, il lavoro di adsorbimento per unità di metilene.
La presenza di un nucleo di idrocarburi nelle micelle consente ai composti insolubili in acqua di dissolversi in soluzioni acquose di tensioattivi, questo fenomeno è chiamato solubilizzazione (quello che si dissolve è un solubilizzato, il tensioattivo è un solubilizzante).
Il fango può essere completamente non polare, può contenere parti sia polari che non polari e sarà orientato come una molecola di tensioattivo.
In ogni caso, durante la solubilizzazione, si verifica un aumento della massa micellare e del numero di aggregazioni non solo per l'inclusione del solubilizzato, ma anche per un aumento del numero di molecole di tensioattivo necessarie per mantenere lo stato di equilibrio.
La solubilizzazione è tanto più efficace quanto minore è il peso molecolare del solubilizzato.

 ~ 72 mN/m2.
~ 72 mN/m2.
 ~ 33 mN/m.
~ 33 mN/m.
L'efficacia dei tensioattivi dipende dalla grandezza della CMC.
Pressione dello strato superficiale 2D

→ -forze di tensione superficiale.
- pressione bidimensionale.
Lo strato superficiale è una forza pari alla differenza tra le tensioni superficiali di una soluzione tensioattiva e di un solvente puro, diretta verso una superficie pulita.
Si stabilisce un equilibrio tra la soluzione e lo strato superficiale



In  c'è una zona dove
c'è una zona dove  linearmente dipendente dalla concentrazione.
linearmente dipendente dalla concentrazione.
G [mol/m2].
 area occupata da una mole di una sostanza
area occupata da una mole di una sostanza
Quindi l'isoterma di pressione bidimensionale avrà la forma 
 è l'isoterma di pressione bidimensionale.
è l'isoterma di pressione bidimensionale.
Dipendenza  da SM:
da SM:

In  - la pressione bidimensionale aumenta bruscamente. In
- la pressione bidimensionale aumenta bruscamente. In  bidimensionale è deformato, il che provoca una forte crescita
bidimensionale è deformato, il che provoca una forte crescita  .
.
Il film su entrambi i lati limitato dalle stesse fasi è chiamato double-face. In tali film si osserva un movimento costante del liquore madre.
I film di spessore inferiore a 5 nm sono chiamati film neri.

Gli strati di adsorbimento dovrebbero avere due caratteristiche: viscosità e facile mobilità, fluidità ed elasticità.
L'effetto Marangoni è autorigenerante.
Triangolo di Gibbs,  - sovrappressione.
- sovrappressione.
Il film è allungato e, poiché parte del liquido è sparito, i tensioattivi si precipitano nello spazio libero. Triangolo di Gibb.
Effetto della forza di adsorbimento dei corpi.
C'è sempre uno strato di adsorbimento sulla superficie del film, per il quale, quindi
Equazione di Langmuir:


 in pressione bidimensionale
in pressione bidimensionale
 - analogo dell'equazione di Shishkovsky
- analogo dell'equazione di Shishkovsky
fenomeni elettrocinetici. Doppio strato elettrico (DES).
modello Helemholtz. Teoria di Gouy-Chapman.
1808 Volo

u – tubo sagomato, immerso in esso 2 elettrodi. La legge dei vasi comunicanti viene violata e c'è un cambiamento nel livello del liquido nel tubo - fenomeni elettrocinetici.
Fenomeni cinetici:
elettroforesi
Elettroosmosi
Potenziale di flusso (flusso).
Potenziale di sedimentazione
1 e 2 sorgono quando viene applicata una differenza di potenziale, 3 e 4 la punzonatura e la sedimentazione di particelle colloidali provocano la comparsa di una differenza di potenziale.
Elettroosmosi è il movimento di un mezzo di dispersione rispetto a una fase dispersa stazionaria sotto l'azione di una corrente elettrica.
elettroforesi è il movimento di particelle della fase dispersa rispetto a un mezzo di dispersione stazionario sotto l'azione di una corrente elettrica.
P  La ragione del verificarsi di fenomeni elettrocinetici è la separazione spaziale delle cariche e la comparsa di un doppio strato elettrico.
La ragione del verificarsi di fenomeni elettrocinetici è la separazione spaziale delle cariche e la comparsa di un doppio strato elettrico.
Il doppio strato elettrico è un condensatore piatto, una piastra è formata da ioni che determinano il potenziale, l'altra da controinoe. Gli ioni sono anche contaminati poiché i coioni che determinano il potenziale vengono spinti nella maggior parte della soluzione. Distanza tra i piatti  . Il potenziale cade linearmente, la differenza di potenziale
. Il potenziale cade linearmente, la differenza di potenziale  .
.
Una differenza di potenziale esterno provoca la comparsa di un modulo di taglio  è una coppia di forze per unità di area che agiscono lungo la superficie di un corpo solido.
è una coppia di forze per unità di area che agiscono lungo la superficie di un corpo solido.

All'equilibrio, il modulo di taglio è uguale al modulo di attrito viscoso (  ).
).

Nelle nostre condizioni  ,
,

 - Equazione di Helemholtz-Smalukovsky
- Equazione di Helemholtz-Smalukovsky
 - spostamento di velocità lineare i fasi.
- spostamento di velocità lineare i fasi.
eè l'intensità del campo elettrico.
 - differenza di potenziale tra le piastre
- differenza di potenziale tra le piastre
 - mobilità elettroforetica [m 2 /(V * s)].
- mobilità elettroforetica [m 2 /(V * s)].
Il modello di Helemholtz non tiene conto del moto termico delle molecole. In realtà la distribuzione degli ioni nel doppio strato è più complessa.
Gouy e Chapman hanno identificato le seguenti cause di DES:
La transizione di uno ione da una fase all'altra quando si stabilisce l'equilibrio.
Ionizzazione di materia in fase solida.
Completamento della superficie da parte degli ioni presenti nel mezzo di dispersione.
Polarizzazione da una sorgente di corrente esterna.
Il doppio strato elettrico ha una struttura sfocata o diffusa. Gli ioni tendono ad essere distribuiti uniformemente in tutto lo strato diffuso.
Lo strato diffuso è costituito da controinioni, la lunghezza dello strato è determinata dalla loro energia cinetica. Ad una temperatura tendente allo zero assoluto, i controinoe sono il più vicino possibile agli ioni potenziali.
Questa teoria si basa su due equazioni:
Equazione di Boltzmann

 - lavorare contro le forze di interazione elettrostatica.
- lavorare contro le forze di interazione elettrostatica.
 è la densità di carica apparente.
è la densità di carica apparente.

Equazione di Poisson 
Poiché lo spessore DEL è molto più piccolo della dimensione delle particelle, e per un DEL piatto, la derivata rispetto alle coordinate  e
e  è abolito.
è abolito. 
Per ey con y<<1 функцию можно разложить в ряд Маклорена:

Ci limitiamo a due membri della serie, quindi:


 - Lo spessore DEL è la distanza alla quale il potenziale DEL diminuisce e una volta.
- Lo spessore DEL è la distanza alla quale il potenziale DEL diminuisce e una volta.
Più bassa è la temperatura, meno  . A Т→0 – DES piatto. Maggiore è la concentrazione, più I, meno
. A Т→0 – DES piatto. Maggiore è la concentrazione, più I, meno  .
.





 “–” significa che il potenziale diminuisce con la distanza. =>
“–” significa che il potenziale diminuisce con la distanza. =>
=>

 ,
,
 - il potenziale diminuisce esponenzialmente.
- il potenziale diminuisce esponenzialmente.
Potenziale di densità di carica superficiale: 
La carica di superficie è una carica spaziale con segno opposto, integrata sulla distanza.




=>


Dove il potenziale diminuisce di 2,7 volte - 
Capacità a doppio strato 
Lo svantaggio della teoria è che la presenza dello strato di Helemholtz non viene presa in considerazione, cioè non tiene conto  , da qui gli errori nella determinazione dei parametri principali. Inoltre non spiega l'effetto di ioni di diversa natura sullo spessore del doppio strato elettrico.
, da qui gli errori nella determinazione dei parametri principali. Inoltre non spiega l'effetto di ioni di diversa natura sullo spessore del doppio strato elettrico.
La teoria di Stern. La struttura di una micella colloidale.
Il doppio strato elettrico è costituito da due parti: densa e diffusa. Uno strato denso si forma come risultato dell'interazione di ioni potenziali con quelli specificamente adsorbiti. Questi ioni, di regola, sono parzialmente o completamente disidratati e possono avere la stessa carica o la carica opposta agli ioni potenziali. Dipende dal rapporto tra l'energia dell'interazione elettrostatica  e potenziale di adsorbimento specifico
e potenziale di adsorbimento specifico  . Gli ioni dello strato denso sono fissi. L'altra parte degli ioni si trova nello strato diffuso; questi ioni sono liberi e possono spostarsi in profondità nella soluzione, ad es. da un'area di maggiore concentrazione a un'area di minore concentrazione. La densità di carica totale è composta da due parti.
. Gli ioni dello strato denso sono fissi. L'altra parte degli ioni si trova nello strato diffuso; questi ioni sono liberi e possono spostarsi in profondità nella soluzione, ad es. da un'area di maggiore concentrazione a un'area di minore concentrazione. La densità di carica totale è composta da due parti.

 - Carica a strati di Helmholtz
- Carica a strati di Helmholtz
 -Carica a strato diffuso
-Carica a strato diffuso
La superficie ha un certo numero di centri di adsorbimento, ognuno dei quali interagisce con un controione. La costante di una tale reazione quasi chimica è:

 , dove
, dove  - frazione molare di controioni in soluzione
- frazione molare di controioni in soluzione
Distribuzione Helmholtz
Il potenziale diminuisce linearmente
Distribuzione potenziale di Gouy. Non c'è uno strato denso, il potenziale diminuisce esponenzialmente dal valore 
Distribuzione severa.
Inizialmente, la potenziale diminuzione è lineare, quindi esponenzialmente.
Quando si applica un campo elettrico nel caso dell'elettroforesi, non è la particella della fase solida che si muove direttamente, ma la particella della fase solida con uno strato di ioni circostanti. DES ripete la forma della particella della fase dispersa. Quando viene applicato un potenziale, una parte dello strato diffuso viene strappata. Viene chiamata la linea di interruzione confine scorrevole.
Viene chiamato il potenziale che sorge al limite di scorrimento a seguito del distacco di una parte dello strato diffuso potenziale elettrocinetico(Potenziale Zeta  ).
).
Viene chiamata una particella di una fase dispersa, con uno strato di controioni che la circondano e un doppio strato elettrico micella.
Regole per scrivere le micelle colloidali:


1-1 elettrolita di carica
T è una particella della fase dispersa.
AA è il confine tra le parti dense e diffuse.
BB è il limite di scorrimento.
Il confine di scorrimento può coincidere o meno con la linea AA.
Viene chiamato il valore del pH a cui il potenziale zeta è zero punto isoelettrico.
CaCl 2 + Na 2 SO 4 → CaSO 4 ↓ + 2NaCl
1. In eccesso di CaCl 2
CaCl 2 ↔ Ca 2+ + 2Cl -
(CaSO 4 m∙nCa 2+ 2( n-x)Cl - ) 2 X + X Cl - - registra le micelle.
CaSO 4 m - aggregato.
CaSO 4 m∙nCa 2+ è il nucleo.
CaSO 4 m∙nCa 2+ 2( n-x)Cl - particella.
2. In eccesso di Na 2 SO 4
Na 2 SO 4 ↔2Na + + SO 4 2-
(CaSO 4 m∙nSO 4 2- 2(n-x)Na + ) 2x- 2xNa + - micella
CaSO 4 m - aggregato.
CaSO 4 m∙nSO 4 2 + è il nucleo.
CaSO 4 m∙nSO 4 2- 2(n-x)Na + - particella
Equazione di Helemholtz-Smoluchowski

 - velocità lineare di spostamento dei confini (in elettroosmosi).
- velocità lineare di spostamento dei confini (in elettroosmosi).
 - differenza di potenziale sulle piastre dei condensatori (in elettroosmosi).
- differenza di potenziale sulle piastre dei condensatori (in elettroosmosi).


 - portata volumetrica della soluzione, Sè l'area della sezione trasversale della cella.
- portata volumetrica della soluzione, Sè l'area della sezione trasversale della cella.

eè l'intensità del campo elettrico.


 (per elettroosmosi).
(per elettroosmosi).
Per il potenziale di flusso:

 - potenziale
- potenziale
 - pressione della membrana
- pressione della membrana
Di norma, il valore delle mobilità elettroforetiche e delle mobilità elettroosmotiche è inferiore a quelle calcolate. Questo è dovuto a:
Effetto di rilassamento (durante il movimento di una particella della fase dispersa, viene violata la simmetria dell'atmosfera ionica).
Frenatura elettroforetica (il verificarsi di ulteriore attrito a seguito del movimento dei controioni).
Distorsione delle linee di flusso nel caso di particelle elettricamente conduttive.
Relazione tra tensione superficiale e potenziale. Equazione di Lippmann.
La formazione di DEL avviene spontaneamente per il desiderio del sistema di ridurne l'energia superficiale. Nel contesto della costanza T e p l'equazione generalizzata del primo e del secondo principio della termodinamica si presenta come:
 (2)
(2)
(3), (1)=(3) =>
=>

 - 1a equazione di Lippmann.
- 1a equazione di Lippmann.
 è la densità di carica superficiale.
è la densità di carica superficiale.
 - capacità differenziale.
- capacità differenziale.
 - 2a equazione di Lippmann.
- 2a equazione di Lippmann.
Insieme a- capacità.
Risolviamo la prima equazione di Lippmann e l'equazione fondamentale di adsorbimento:
 ,
,


 , poi
, poi
 - Equazione di Nernst
- Equazione di Nernst
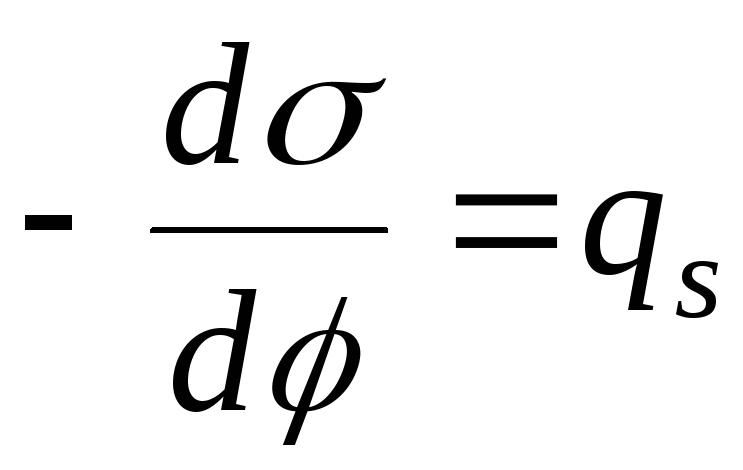 ,
,
 ,
,




 - l'equazione della curva elettrocapillare (ECC).
- l'equazione della curva elettrocapillare (ECC).
A  :
: , ma
, ma 
I tensioattivi cationici (CSAS) riducono il ramo catodico dell'ECC.
I tensioattivi anionici (ASS) riducono il ramo anodico dell'ECC.
I tensioattivi non ionici (NSA) riducono la parte centrale dell'ECC.
Stabilità dei sistemi dispersi. Pressione di incuneamento.
I sistemi dispersi possono essere suddivisi:
I sistemi termodinamicamente instabili possono essere cineticamente stabili a causa del passaggio a uno stato metastabile.
Esistono due tipi di stabilità:
Stabilità della sedimentazione (rispetto alla gravità).
Stabilità aggregativa. (in relazione all'attaccamento)
Coagulazioneè il processo delle particelle che si uniscono, portando alla perdita di stabilità aggregativa. La coagulazione può essere causata da variazioni di temperatura, pH, agitazione, ultrasuoni.
Distinguere la coagulazione:
Reversibile.
Irreversibile.
La coagulazione procede con l'introduzione di elettroliti.
Regole di coagulazione:

Film- Questa è la parte del sistema situata tra due interfacce.
pressione disgiunta si verifica con una forte diminuzione dello spessore del film a causa dell'interazione degli strati superficiali in avvicinamento.

«-» - quando lo spessore del film diminuisce, la pressione di disgiunzione aumenta.
P 0 è la pressione nella fase di massa, che è una continuazione dell'intercalare.
P 1 è la pressione nel film.
Teoria della stabilità. DLFO (Deryagin, Landau, Fairway, Overbeck).
Secondo la teoria DLVO, nella pressione disgiunta si distinguono due componenti:
elettrostatico PE (positivo, è dovuto alle forze di repulsione elettrostatica). Corrisponde a una diminuzione dell'energia di Gibbs con l'aumento dello spessore del film.
Molecolare PM (negativo, per l'azione di forze attrattive). È causato dalla compressione del film dovuta alle forze chimiche superficiali, il raggio d'azione delle forze è di decimi di nm con un'energia dell'ordine di 400 kJ/mol.
Energia di interazione totale:

 - il sistema è aggregato stabile
- il sistema è aggregato stabile
 - sistema instabile
- sistema instabile
P  componente positiva.
componente positiva.
L'aumento è dovuto all'aumento dell'energia potenziale durante la compressione di film sottili. Per i film spessi, l'energia ionica in eccesso viene compensata ed è uguale all'interazione energetica nella massa del mezzo di dispersione.
Se un  (
( - spessore del film,
- spessore del film,  - raggio dello ione) l'assottigliamento del film porta alla scomparsa e riduzione in esso di molecole e ioni con un'energia superficiale minima. Il numero di particelle vicine diminuisce, di conseguenza aumenta l'energia potenziale delle particelle che rimangono nel film.
- raggio dello ione) l'assottigliamento del film porta alla scomparsa e riduzione in esso di molecole e ioni con un'energia superficiale minima. Il numero di particelle vicine diminuisce, di conseguenza aumenta l'energia potenziale delle particelle che rimangono nel film.


La teoria DLVO considera l'interazione delle particelle come l'interazione delle piastre.

Le particelle non interagiscono



 - Equazione di Laplace,
- Equazione di Laplace,  ,
,


Per superfici debolmente cariche
Per superfici molto cariche:

La componente molecolare è l'interazione di due atomi:
 ~
~
Interazione di un atomo con una superficie:

Prendiamo due record:
D  Per ottenere la componente molecolare è necessario sommare tutte le energie di interazione degli atomi delle piastre destra e sinistra.
Per ottenere la componente molecolare è necessario sommare tutte le energie di interazione degli atomi delle piastre destra e sinistra.
dove  - Costante di Hamaker (tiene conto della natura dei corpi interagenti).
- Costante di Hamaker (tiene conto della natura dei corpi interagenti).
Quella. l'energia di interazione delle particelle in un sistema può essere espressa utilizzando curve di potenziale.
I è il minimo potenziale primario. Questa è una zona di coagulazione irreversibile, prevalgono le forze di attrazione.
II - zona di stabilità aggregativa, prevalgono le forze repulsive.
III - minimo potenziale secondario (o zona di flocculazione). Tra le particelle della fase dispersa c'è uno strato di elettrolita e le particelle possono essere separate e trasferite nella zona di stabilità aggregativa.

Curva 1: il sistema è aggregativamente stabile.
La curva 2 è stabile nella zona I, non stabile nella zona II.
Curva 3 - si è verificata coagulazione nel sistema.
Curva 4 - al punto 4, l'energia totale di interazione U=0,  , questo punto estremo corrisponde all'inizio di una rapida coagulazione.
, questo punto estremo corrisponde all'inizio di una rapida coagulazione.
Ci sono due casi:
1. Le superfici sono debolmente cariche:
U \u003d U E + U M \u003d 0
 (1)
(1)
2)

 (2)
(2)



 - questo è lo spessore dello strato corrispondente all'inizio del processo di coagulazione.
- questo è lo spessore dello strato corrispondente all'inizio del processo di coagulazione.






 - per superfici debolmente cariche
- per superfici debolmente cariche
 poi
poi 
2. Per superfici molto cariche:
 (1)
(1)
2)

 (2)
(2)


 (3)
(3)
 ,
,
Facciamo quadrato (3)

Coagulazione:
Nell'adsorbimento specifico, gli ioni possono essere adsorbiti in una quantità superequivalente in modo tale che la superficie possa cambiare la sua carica. La superficie è in ricarica.
Nel caso di adsorbimento specifico, possono essere adsorbiti non solo ioni di segno opposto, ma anche di uno ione.
Se vengono adsorbiti ioni dello stesso segno della superficie, nello strato superficiale non ci sarà un calo del potenziale, ma la sua crescita.

Coagulazione di neutralizzazione (si verifica con la partecipazione di particelle debolmente cariche e dipende non solo dalla carica dell'elettrolita coagulante, ma anche dal potenziale al confine degli strati densi e diffusi).
La teoria della coagulazione veloce di Smoluchowski.

Dipendenza della velocità di coagulazione dalla concentrazione di elettroliti.
I – il tasso di coagulazione è basso,
II - la velocità di coagulazione è praticamente proporzionale alla concentrazione di elettroliti.
III - l'area di rapida coagulazione, la velocità è praticamente indipendente dalla concentrazione.
Punti chiave:
Il sol iniziale è monodisperso, particelle simili hanno una forma sferica.
Tutte le collisioni di particelle sono efficaci.
Quando due particelle primarie si scontrano, si forma una particella secondaria. Secondaria + primaria = terziaria. Primario, secondario, terziario - molteplicità.
In termini di cinetica chimica, il processo di coagulazione può essere descritto dall'equazione:

La soluzione sarà l'equazione: 
 - tempo di semicoagulazione. Questo è il tempo durante il quale il numero di particelle di sol diminuisce di 2 volte.
- tempo di semicoagulazione. Questo è il tempo durante il quale il numero di particelle di sol diminuisce di 2 volte.

 ,
,
 ,
,
 ,
,


All'aumentare della molteplicità, il massimo delle curve di coagulazione si sposta verso valori maggiori  .
.
Svantaggi:
Assunzione di monodispersione.
L'ipotesi sull'efficacia di tutte le collisioni.
Pagina corrente: 6 (il libro ha un totale di 19 pagine) [estratto di lettura accessibile: 13 pagine]
Font:
100% +
34. Natura delle forze di adsorbimento
L'interazione tra le molecole dell'adsorbente con la superficie dell'adsorbente al cosiddetto. l'assorbimento fisico può essere dovuto a vari motivi. Quindi il potenziale che determina l'interazione di una molecola adsorbente con un atomo di un adsorbente non polare può essere espresso come segue:
θ = −Cr 6 +fr 12 ,
dove r è la distanza tra i centri delle particelle; C è la costante di attrazione della dispersione; B è una costante che caratterizza l'energia delle forze repulsive.
È abbastanza ovvio che a distanze relativamente lontane dovrebbero prevalere le forze attrattive e, a distanze ravvicinate, le forze repulsive. Inoltre, a determinate distanze, queste forze devono essere uguali, il che corrisponderà all'energia libera minima. Ma è importante notare che durante l'adsorbimento, le forze di dispersione agiscono simultaneamente tra ciascuna particella non polare.
Poiché l'energia di interazione delle particelle può diminuire rapidamente con la distanza, è sufficiente eseguire la somma degli atomi adsorbenti più vicini per determinare il potenziale delle forze di adsorbimento. È importante che, nel caso di adsorbimento di molecole complesse non polari, l'energia potenziale possa essere approssimativamente calcolata come la somma di tutte le energie potenziali di adsorbimento delle unità della molecola.
Se l'adsorbente è costituito da ioni, l'azione delle forze di dispersione già note può essere integrata dall'azione delle forze di attrazione di induzione dei dipoli, che sono indotte nelle molecole dell'adsorbente da un campo elettrico, che, a sua volta, viene creato dagli ioni del reticolo adsorbente.
Con tale interazione, la quota delle forze induttive nell'interazione di adsorbimento può essere proporzionale alla polarizzabilità della molecola di adsorbimento e al quadrato dell'intensità del campo su questa superficie adsorbente.
Se, d'altra parte, le molecole adsorbenti polari vengono adsorbite su un adsorbente polare, i dipoli in questo caso polarizzano gli atomi dell'adsorbente, cioè come se in essi inducessero momenti elettrici. A causa di questa influenza, l'interazione induttiva si aggiunge a quella di dispersione.
L'interazione induttiva stessa è generalmente piccola e, a seconda del dipolo della molecola adsorbente e della polarizzabilità dell'adsorbente, può raggiungere valori elevati. Nel caso in cui le molecole vengano adsorbite su un adsorbente che ha ioni o dipoli sulla superficie, un cosiddetto. interazione di ioni o dipoli dell'adsorbente con il campo elettrostatico dell'adsorbente stesso.
In questo caso, le molecole adsorbenti possono anche orientarsi nel campo dell'adsorbente e si verifica un'interazione orientativa di Coulomb. Di solito accade che le energie delle interazioni induttive e orientative siano inferiori all'energia dell'interazione di dispersione, e quindi si presume che l'energia di attrazione intermolecolare sia determinata dall'energia di attrazione di dispersione.
Inoltre, la formazione di un legame idrogeno può servire come causa di adsorbimento. Un legame di questo tipo può sorgere durante l'adsorbimento su adsorbenti che contengono gruppi idrossilici di molecole come acqua, alcoli, ammoniaca e ammine sulla superficie. Quando si forma un legame idrogeno, l'energia di interazione dell'adsorbente con l'adsorbente può essere piuttosto grande e il calore che viene rilasciato durante tale adsorbimento è molto maggiore del calore di adsorbimento di sostanze simili per forma e dimensione delle molecole, ma non formano un legame idrogeno.
È importante notare che, conoscendo la descrizione termodinamica dello strato superficiale al confine "adsorbente - adsorbente", la sua struttura, la natura dei vari tipi di forze, la dinamica del processo, si può procedere allo studio di più complessi processi di adsorbimento.
35. Adsorbimento come concentrazione spontanea sull'interfaccia di fase di sostanze che riducono la tensione interfacciale
I tensioattivi si dividono in due grandi gruppi: attivo e inattivo sostanze.
I tensioattivi possono accumularsi nello strato superficiale e in questo caso si verifica un adsorbimento positivo. G > 0.
Tali tipi di sostanze devono avere una tensione superficiale che, a sua volta, deve essere inferiore alla tensione superficiale del solvente, altrimenti l'accumulo della sostanza nello strato superficiale sarebbe sfavorevole e deve avere una solubilità relativamente bassa. Con una solubilità sufficientemente buona, le molecole di tensioattivo tendono a lasciare la superficie in profondità nella soluzione. Pertanto, i tensioattivi verranno preferibilmente spinti fuori dalla massa del liquido verso la superficie.
Ma con l'accumulo di sostanze al confine della soluzione nelle molecole di queste sostanze, che interagiscono debolmente tra loro, l'interazione intermolecolare nello strato superficiale diminuirà e la tensione superficiale diminuirà.
Tensioattivi rispetto allo strato d'acqua ci sono molti tipi di composti organici, acidi grassi con un radicale idrocarburico sufficientemente grande, sali di questi acidi (saponi), acidi solfonici e loro sali, nonché vari tipi di alcoli e ammine. Una caratteristica della maggior parte delle molecole è la loro anfifilia: la molecola è costituita da due parti di un gruppo polare e un radicale idrocarburico non polare. Possedere un momento di dipolo significativo e un gruppo polare ben idratante può determinare l'affinità del tensioattivo per l'ambiente acquoso. Ma il radicale idrocarburico è la causa che abbassa la solubilità di questi composti.
Tensioattivi inattivi di superficie- questo tipo di sostanze, tendenti a lasciare la superficie del liquido nel suo volume, di conseguenza, il cosiddetto. adsorbimento negativo G < 0. Поверностно-инактивные вещества также обладают значительным поверхностным натяжением, значительно большим, чем натяжение у растворителя (иначе эти вещества способны самопроизвольно накапливаться в поверхностном слое), также обладают высокой растворимостью, что способствует их стремлению уйти с поверхности жидкости в объем. Взаимодействие между молекулами поверхностно-инактивного вещества и растворителя всегда больше, чем взаимодействие между самими молекулами растворителя, поэтому они и стремятся перейти в объем раствора. Sostanze inattive di superficie rispetto all'acqua sono molti gli elettroliti inorganici: acidi, alcali, sali. Le molecole di sostanze inattive in superficie non hanno una parte idrofobica e possono decomporsi in acqua in ioni altamente idratanti.
Esempi sostanze tensioattive sono anche alcuni composti organici in cui la parte non polare della molecola è assente o molto piccola. Queste sostanze includono acidi formico e aminoacetico.
Nei solventi non acquosi, gli elettroliti inorganici sono anche in grado di aumentare la tensione superficiale, e questo dipende dal solvente.
Per esempio, quando lo ioduro di sodio viene introdotto nel metanolo, la tensione superficiale aumenta notevolmente; per l'etanolo, la tensione superficiale è circa 2 volte maggiore. L'attività superficiale delle sostanze può dipendere non solo dalla natura della sostanza, ma anche dalle proprietà del solvente. Se un solvente ha un'elevata tensione superficiale, questo soluto può mostrare un'attività superficiale significativa.
36. Teorie dell'adsorbimento
Consideriamo le teorie di adsorbimento più comuni che descrivono i singoli tipi di adsorbimento sull'interfaccia "gas solido" o "soluzione solida".
La teoria dell'adsorbimento monomolecolare di I. Langmuir.
1. L'adsorbimento è localizzato ed è causato da forze vicine a quelle chimiche.
2. L'adsorbimento avviene solo su centri attivi - sporgenze o depressioni sulla superficie dell'adsorbente, caratterizzati dalla presenza di valenze libere. I centri attivi sono considerati indipendenti e identici.
3. Ciascun centro attivo è in grado di interagire con una sola molecola di adsorbato; sulla superficie si può formare solo uno strato di molecole adsorbite.
4. Il processo di adsorbimento è reversibile ed equilibrato; la molecola adsorbita viene trattenuta dal centro attivo per qualche tempo, dopodiché viene desorbita; Dopo qualche tempo si stabilisce un equilibrio dinamico.
Il massimo valore di adsorbimento possibile G o si ottiene a condizione che tutti i centri attivi siano occupati da molecole di adsorbato. Equazione dell'isoterma di adsorbimento monomolecolare relativa al valore di adsorbimento G con concentrazione di adsorbato Insieme a, sembra:
dove b- una costante per una data coppia di valore "adsorbente - adsorbato" (il rapporto tra le costanti di velocità di desorbimento e adsorbimento), numericamente uguale alla concentrazione dell'adsorbato, alla quale è occupata la metà dei centri attivi.

Il grafico dell'isoterma di adsorbimento di Langmuir è mostrato nella Figura 2. La costante b definiamo graficamente disegnando una tangente all'isoterma di adsorbimento nel punto Insieme a= 0. Quando si descrive il processo di adsorbimento dei gas nell'equazione, la concentrazione può essere sostituita da un valore proporzionale della pressione parziale. Teoria dell'adsorbimento monomolecolare I. Langmuir applicabile per descrivere i processi di adsorbimento di gas e sostanze disciolte a basse pressioni (concentrazioni) dell'adsorbato.
La teoria di Polanyi dell'adsorbimento polimolecolare descrive le isoterme di adsorbimento a forma di S, la cui forma indica la possibile interazione delle molecole adsorbite con l'adsorbato.
1. L'assorbimento è causato da forze fisiche.
2. La superficie dell'adsorbente è omogenea, non sono presenti centri attivi; le forze di adsorbimento formano un campo di forza continuo vicino alla superficie dell'adsorbente.
3. Le forze di adsorbimento agiscono a una distanza maggiore della dimensione della molecola di adsorbato, cioè c'è un certo volume di adsorbimento vicino alla superficie dell'adsorbente, che viene riempito di molecole di adsorbato durante l'adsorbimento.
4. L'attrazione di una molecola di adsorbato da parte della superficie adsorbente non dipende dalla presenza di altre molecole nel volume di adsorbimento, per cui è possibile l'adsorbimento polimolecolare.
5. Le forze di adsorbimento non dipendono dalla temperatura e, pertanto, al variare della temperatura, il volume di adsorbimento non cambia.
Equazione di Freundlich. La superficie dell'adsorbente è disomogenea, si verifica un'interazione tra le particelle adsorbite, i centri attivi non sono completamente indipendenti l'uno dall'altro. G. Freindlich ha suggerito che il numero di moli di gas o soluto adsorbito per unità di massa dell'adsorbente (il cosiddetto adsorbimento specifico X/m), deve essere proporzionale alla pressione di equilibrio (per i gas) o alla concentrazione di equilibrio (per le sostanze adsorbite dalla soluzione) dell'adsorbente elevato ad una certa potenza, che è sempre inferiore all'unità:
X / m = aP n X / m = corrente alternata n.
esponenti n e fattore di proporzionalità un determinato sperimentalmente.
37. Termodinamica del processo di adsorbimento. Equazione di adsorbimento di Gibbs
Per studiare il fenomeno dell'adsorbimento al confine "soluzione - gas" è necessario stabilire una relazione tra l'eccesso di sostanza adsorbita nello strato superficiale ( G), concentrazione di tensioattivo in soluzione ( insieme a) e tensione superficiale ( σ ) al confine di fase “soluzione-gas”. È più opportuno considerare i fenomeni dal punto di vista termodinamico e mettere in relazione l'adsorbimento di un soluto con una variazione dell'energia libera della superficie o della sua tensione superficiale. Questa connessione è stata effettuata W. Gibbs in 1876, che è stato nominato "Equazione di adsorbimento di Gibbs":
G = – insieme a / RT X dσ/dc.
Puoi ancora immaginare Equazione di Gibbs, basato sulla termodinamica, utilizzando il potenziale isobarico-isotermico G, potenziali chimici μ 1 e μ 2 , e anche usando n 1 e n 2 il numero di moli dei componenti. Dopo averlo analizzato tenendo conto dell'entropia S, volume V e pressione P, possiamo scrivere la seguente equazione:
dG=– SDT+VdP+σds+ μ 1 giorno n 1 + μ 2 giorni 2 .
Lo uguagliamo a zero e, tenendo conto della temperatura e della pressione costanti, si semplifica in un'equazione della forma:
sd σ + n 1 g μ 1 + n2d μ 1 = 0.
Tenendo conto del fatto che per soluzioni diluite il potenziale chimico del secondo componente è espresso come segue:
μ 2 = μ 2 0 +RT ln c,
e dato che la temperatura è costante
dμ 2 =rtdnc,
sostituendo questa equazione in
![]()
otteniamo l'equazione di adsorbimento di Gibbs desiderata. Sulla base dell'equazione, si può vedere che se la tensione superficiale σ aumenta con la concentrazione insieme a, allora la concentrazione della sostanza disciolta sullo strato superficiale è inferiore al volume della soluzione (il cosiddetto adsorbimento negativo) e se la tensione superficiale σ diminuisce con l'aumentare della concentrazione insieme a, allora la concentrazione nello strato è maggiore che nel volume (adsorbimento positivo) e, infine, se σ non dipende da insieme a, quindi la concentrazione della sostanza nello strato in superficie e nel volume è la stessa. L'equazione di Gibbs è stata derivata utilizzando la termodinamica. È difficile verificare in pratica questa equazione, a causa della complessità di determinare la concentrazione di una sostanza disciolta in una superficie stratificata. Esperto B. McBen trovato che uno strato molto sottile di liquido è stato tagliato dalla superficie della soluzione utilizzando il dispositivo. Un'ulteriore determinazione di tutti i parametri dell'equazione di Gibbs ha mostrato che i valori di adsorbimento trovati sperimentalmente coincidevano con i valori calcolati utilizzando l'equazione di Gibbs all'interno dell'errore sperimentale. A causa dell'omogeneità e della levigatezza della superficie di qualsiasi liquido, quando si studia l'adsorbimento sulla sua superficie, le solite idee sui centri attivi sono completamente inapplicabili. Alla temperatura critica, la differenza tra le fasi adiacenti scompare e la tensione superficiale, di regola, diventa uguale a zero. L'adsorbimento di gas e vapori ha una così ampia applicazione pratica che in letteratura, soprattutto in quella tecnica, si può ritrovare questo concetto, che viene utilizzato solo in relazione a processi sulla superficie dei solidi.
Questo concetto, così come i modelli più generali di adsorbimento, come l'equazione di Gibbs considerata, è applicabile a tutti i limiti di fase. Utilizzando l'equazione di Gibbs e tutte le disposizioni da essa derivanti, determinato il valore di Г, è possibile costruire un'isoterma di adsorbimento.
38. Peculiarità dell'adsorbimento su materiali microporosi. La teoria del potenziale di Polan. Potenziale di adsorbimento
La teoria di Glade considera l'adsorbimento fisico non localizzato, che è direttamente dovuto alle forze di van der Waals tra l'adsorbente e l'adsorbato (questa può essere considerata la prima posizione). La seconda posizione di questa teoria è il concetto di campo di forza (o potenziale) dell'adsorbente, che si estende per una notevole distanza dalla superficie; lo strato di adsorbimento che appare in questo campo è polimolecolare. Se consideriamo l'adsorbimento dei gas, la densità di questo strato diminuisce lungo una certa normale dalla superficie. Se consideriamo l'adsorbimento del vapore, sulla superficie si forma uno strato liquido di un certo spessore. Il campo nella teoria di Polanyi è considerato come una serie di superfici equipotenziali, ogni superficie corrisponde ad un certo valore del potenziale ε , e ogni superficie successiva sarà più piccola della precedente. Ciascuna di queste superfici nello spazio ritaglia strati di un certo volume, designati come v io. Il compito della teoria di Polanyi è trovare il passaggio dalle coordinate usuali dell'isoterma ( x, pag) per inserire i parametri ε i e v io, con ulteriore determinazione del collegamento tra questi parametri principali. La prima parte del problema, che Polanyi ha esposto, è piuttosto complicata e in molti casi non può avere soluzioni definite, ma per il caso dell'adsorbimento del vapore, questa parte del problema è risolta in prima approssimazione in modo molto semplice. Per uno strato di adsorbimento liquido, la parte riempita del volume sarà uguale a:
v io \u003d x (M / d),
dove dè la densità della sostanza allo stato liquido.
Nella sua teoria, M. Polyany introduce un'altra disposizione sull'assenza del cosiddetto. screening sul campo nel processo di adsorbimento, il valore ε in questa teoria dello spazio è un valore costante (qualcosa come un potenziale gravitazionale) indipendentemente dal fatto che alcune molecole di adsorbato esistano tra un dato punto e una superficie solida, o che tutto lo spazio sia libero. Polyani introduce il concetto potenziale di adsorbimento ε , che è il lavoro isotermico di compressione del vapore quando viene trasferito dalla pressione di equilibrio R nella fase bulk lontano dalla superficie nella regione dello strato superficiale con pressione di vapore saturo p 0 quindi l'espressione per determinare il potenziale sarà simile a:
ε = RT ln R 0 / R.
Con l'aiuto di tale equazione, si può passare dalle coordinate x, p alle coordinate ε e v e ottieni una curva, che viene chiamata "caratteristica". Polanyi scoprì nei suoi esperimenti che tali curve, costruite dai dati sperimentali delle isoterme ottenute, hanno la seguente proprietà: sono invarianti rispetto a T, ovvero, tutte le curve di questo tipo possono giacere su una curva ε −ε .
M. Polyany ha accettato questa posizione come postulato, cioè:
Questa proprietà di Polyani è di grande importanza pratica; può costruire una famiglia di isoterme da un'isoterma di adsorbimento sperimentale.
La teoria di Polanyi non fornisce un'espressione analitica per l'isoterma o la funzione del potenziale rispetto al volume, ma consente di calcolare la coordinata per una data temperatura se almeno un'isoterma è nota. Questo risultato è molto importante per i calcoli tecnologici, perché per gas simili sullo stesso adsorbente, le curve di adsorbimento possono risultare vicine tra loro e in molti casi possono essere sovrapposte.
39. Curva caratteristica di adsorbimento. Invarianza termica e affinità delle curve caratteristiche
Il campo di forza che appare sulla superficie dell'adsorbente può essere simile per molti aspetti al campo gravitazionale. Nel campo di adsorbimento possono essere rappresentate superfici di potenziale, cioè superfici per le quali è caratteristico lo stesso potenziale di adsorbimento. Sotto il concetto di potenziale di adsorbimento θ dovrebbe essere inteso come nient'altro che il lavoro svolto contro le forze di adsorbimento spostando 1 mole di adsorbato da un certo punto del campo a una determinata fase gassosa. Il massimo potenziale di adsorbimento esisterà al confine "adsorbente - volume di adsorbimento". Ma al confine "volume - fase gassosa" (qui termina l'azione delle forze di adsorbimento), il potenziale di adsorbimento deve essere uguale a zero. La variazione del potenziale di adsorbimento con una variazione del volume di adsorbimento può essere rappresentata sotto forma di curve. Questo è stato fatto per la prima volta da M. Polyani. Tali tipi di curve non dipendono dalla temperatura e possono essere caratteristici di ogni specifico adsorbente; tali tipi di curve sono generalmente chiamati curve caratteristiche di adsorbimento. La teoria dell'adsorbimento polimolecolare presuppone che l'equazione di stato del gas si applichi alla quantità di adsorbimento. Di conseguenza, le isoterme che caratterizzano la dipendenza della densità dell'adsorbato dal volume per diverse temperature assomigliano alle isoterme della dipendenza della pressione dal volume. A basse temperature, le forze di adsorbimento sulla superficie possono far condensare il vapore in un liquido di una certa densità. A temperature inferiori a quella critica, durante la condensazione, l'intero volume di adsorbimento sarà riempito di liquido. In questo caso, la curva di adsorbimento sarà quasi parallela all'asse delle ascisse, il che è correlato alla bassa comprimibilità del liquido. Quindi la curva di adsorbimento al confine "volume - fase gassosa" scende bruscamente e, di conseguenza, la densità dell'adsorbato raggiunge il valore di una certa densità della fase gassosa. A temperature superiori a quella critica, l'adsorbente può comportarsi come un gas ideale, e il grafico sarà espresso come un'isoterma della dipendenza per un gas ideale, a condizione che pv = RT. In tali condizioni, il gas adsorbito avrà una densità massima sulla superficie stessa dell'adsorbente e una densità minima nelle immediate vicinanze della fase gassosa. Inoltre, in questo caso è importante notare che la densità dell'adsorbato nello strato di adsorbimento non raggiunge da nessuna parte la densità del liquido stesso. E se la temperatura è molto vicina a quella critica, la dipendenza della densità dal volume sarà espressa da una curva simile nell'aspetto all'isoterma, che è descritta Equazione di van der Waals. In questo scenario, parte della sostanza adsorbita sarà nel volume adsorbito allo stato liquido e parte della sostanza adsorbita sarà allo stato gassoso. Quindi la curva diminuirà più nettamente nella sezione che corrisponde al passaggio dal liquido al gas. Se si costruisce una curva caratteristica dall'isoterma di adsorbimento sperimentale di uno degli adsorbenti, e conoscendo i corrispondenti coefficienti di affinità per qualche altro adsorbente, si può trovare l'isoterma di adsorbimento e costruirla per un altro adsorbente. La teoria del potenziale di adsorbimento permette di calcolare diverse isoterme di adsorbimento di diversi vapori sullo stesso adsorbente, inoltre, secondo la curva caratteristica, che si ottiene dall'isoterma di adsorbimento di un vapore, poiché il rapporto del potenziale di adsorbimento non dipende sui volumi di adsorbimento.
affinità(dal latino affinis - "correlato") - cromatografia di affinità. Il metodo di purificazione e separazione delle proteine si basa sulla loro interazione selettiva con un ligando legato covalentemente a un vettore inerte (cromatografia di affinità). La misurazione dell'affinità di un tossico per un recettore è, infatti, uno studio sperimentale della relazione tra la quantità di una sostanza aggiunta al mezzo di incubazione e la quantità del complesso tossico-recettore formatosi a seguito dell'interazione.
RIFERIMENTI
1. Pope MT, Müller A. Chimica del poliossometallato: un vecchio campo con nuove dimensioni in diverse discipline // Angew. Chimica. int. ed. inglese - 1991. - V. 30. - P. 34-48.
2. Pop MS Eteropoli e isopolimetallati. - Novosibirsk: Nauka, 1990. - 232 pag.
3. Maksimov G.M. Risultati nel campo della sintesi dei poliossometallati e dello studio degli eteropoliacidi // Uspekhi khimii. - 1995. - T 64. - N. 5. - S. 480-493.
4. Dobrynina NA Composti isopoli ed eteropoliti // Journal of Inorganic Chemistry. - 2002. - T. 47. - N. 4. - S. 577-587.
5. Cartmell E., Fowles GWA Valenza e struttura delle molecole. -M.: Chimica, 1979. - S. 272.
6. Fedotov MA, Samokhvalova E.P., Kazansky L.P. Spostamenti diamagnetici e paramagnetici NMR 17O e 183W in eterodecatungstati XW10O36- (X=Ln, Th, U) in soluzioni acquose // Poliedro. -1996. - V. 15. - N. 19. - P. 3341-3351.
7. Shiozaki R., Inagaki A., Ozaki A., Kominami H., Yamaguchi S., Ichiha-ra J., Kera Y. Comportamento catalitico di serie di lantanidi decatungstati per H2O2 - ossidazioni di alcoli e olefine. Alcuni effetti chimici del 4fn - elettrone nello ione lantanide(III) sui catalizzatori // J. Alloys Compounds. -1997. - V. 261. - P. 132-139.
8. Kazansky L.P., Golubev AM, Baburina I.I., Torchenko-va E.A., Spitsyn V.I. Spettri vibrazionali di eteropoli-
anioni XW10O36n- // Izvestiya AN SSSR. Ser. chimica. - 1978. - N. 10. - S. 2215-2219.
9. Kolenkova MA, Kerin O.E. Metallurgia dei metalli rari sparsi e leggeri. - M.: Metallurgia, 1977. - S. 12.
10. Kaziev G.Z., Dutov A.A., Olgin K.S., Belsky V.K., Zavod-nik V.E., Hernandez-Peres T., Kanaev A.A. Sintesi e studio della diffrazione dei raggi X del deamolibdenodicobaltato di potassio (III) // Journal of Inorganic Chemistry. - 2004. - T. 49. - N. 5.
11. Chimica e tecnologia degli elementi rari e in tracce, ed. K.A. Bolshakov. - M.: Scuola Superiore, 1976. - Parte 2. -S. 166, 174; Parte 3. - S. 176, 233, 170, 171, 228.
12. Zagabria P.A., Borzenko M.I., Vasiliev S.Yu., Tsirlina G.A. Cinetica dell'elettroriduzione dello ione centrale nel cerio(GU)-decatungstato // Elektrokhimiya. - 2004. - T. 40. - N. 5. - S. 565-575.
13. Safronov S.M., Berezina E.M., Terent'eva G.A., Chernov E.B., Filimoshkin A.G. Estrapolazione non lineare delle dipendenze dalla concentrazione della viscosità ridotta e dalla struttura delle soluzioni polimeriche. -2001. - ser. B. - T. 43. - N. 4. - S. 751-754.
14. Romanova TA, Krasnov PO, Kachin S.V., Avramov P.V. Teoria e pratica della modellazione al computer di nanooggetti.
Krasnoyarsk: IPT KSTU, 2002. - 223 p.
UDC 544.3:622.331
TERMODINAMICA DELL'ADSORBIMENTO DI COMPOSTI SU ACIDI UMICI
SG Maslov, LI Tarnovskaja
E-mail del Politecnico di Tomsk: [email protetta]
Riassunto—È stato studiato il processo di adsorbimento di composti organici (n-alcani, cicloalcani, alcheni, eteri, esteri ed eteri ciclici, chetoni, alcoli, idrocarburi aromatici e cloro-sostituiti) su acidi umici della torba originale e trattata termicamente mediante il metodo gascromatografico per determinare i parametri di adsorbimento e termodinamici. La caratterizzazione degli acidi umici è data da metodi generalmente accettati nella chimica dei combustibili fossili solidi mediante spettroscopia NMR.
Vengono rivelate le relazioni tra caratteristiche fisico-chimiche e parametri di ritenzione sull'adsorbente. Sono state stabilite differenze nei processi di adsorbimento sugli acidi umici della torba originale e trattata termicamente a causa dell'aumento del contenuto di gruppi contenenti ossigeno e frammenti aromatici nei campioni trattati termicamente. Viene mostrata la relazione tra la probabilità termodinamica del processo di adsorbimento sugli acidi umici e la polarità degli adsorbati.
introduzione
Le informazioni in letteratura sulle proprietà di adsorbimento degli acidi umici (HA) sono chiaramente insufficienti. Il punto di vista tradizionale che il processo di adsorbimento, da un lato, sia di natura volumetrica e, dall'altro, sia specifico per la presenza di vari gruppi funzionali, non dà un'idea chiara del meccanismo di questo fenomeno. Ci sono prove che i nuclei aromatici condensati possono anche essere portatori di proprietà di adsorbimento. Va notato che la maggior parte degli autori ha studiato il processo di adsorbimento di ioni metallici e sostanze inorganiche su HA. Ci sono pochissimi lavori dedicati allo studio della capacità di adsorbimento dell'HA rispetto ai composti organici e non sono sistematici.
Lo scopo di questo lavoro è studiare la capacità di adsorbimento dell'HA rispetto a un certo numero di composti organici.
Esperimento e metodo
Come oggetto di studio è stata utilizzata la torba di carice con un grado di decomposizione del 35% del deposito di Tagansky della regione di Tomsk.
L'HA è stato ottenuto secondo la prescrizione del Peat Institute e caratterizzato sia dai metodi generalmente accettati nella chimica dei combustibili fossili solidi sia dal metodo della spettroscopia NMR. Le proprietà di adsorbimento dell'HA sono state studiate con un metodo gascromatografico modificato.
Lo studio è stato condotto su un cromatografo Tsvet-100 con un rivelatore di conducibilità termica a
uso dell'elio come gas di trasporto. Il cromatografo è dotato di un manometro di riferimento per misurare il gradiente di pressione della colonna e correggere la compressibilità. I campioni di HA sono stati macinati in un mortaio di agata e la frazione
0,5...0,25 mm. Una colonna di acciaio lunga 1 m e di 4,2 mm di diametro è stata riempita con HA preparati in una quantità di 6,7 g ad una pressione di 10-3 Pa. I campioni sono stati riscaldati in modalità lineare da 333 a 363 K ad una velocità di 2 gradi/min. Come adsorbati sono stati utilizzati composti organici: alcani, cicloalcani, alcani sostituiti con cloro, areni, alcoli, alcheni, chetoni, eteri semplici, complessi e ciclici. I campioni analizzati sono stati iniettati nel cromatografo utilizzando una microsiringa.
Tabella 1. Caratteristiche della torba di carice, peso. %
Composizione tecnica ed elementare Composizione del gruppo per massa organica
No Aa V1e "S^e" H1e" LG B VRV LGV GK FK C NO
6,7 7,9 68,4 53,4 5,9 2,1 38,5 2,6 1,8 30,9 35,1 11,2 2,1 15,5
Nota: № - umidità analitica; A1 - contenuto di ceneri per combustibile secco; V11" - contenuto volatile sulla massa combustibile; B - bitume; VRV e LGV - sostanze idrosolubili e facilmente idrolizzabili; FA - acidi fulvici; C - cellulosa; HO - residuo non idrolizzabile
Tabella 2. Caratteristiche degli acidi umici della torba
Torba HA Composizione elementare, % Contenuto, mg.eq./g
Peso Atomico Rapporti atomici □= o o C COOH, OH,
S1e" N1e" KOLZ" S N O/S n/s
Baseline 54,84 6,66 35,60 35,28 48,41 15,84 0,45 1,37 2,56 6,00
Termotrattato 60,09 5,22 34,69 40,48 41,90 17,01 0,42 1,03 3,06 6,89
Tabella 3. Contenuto di frammenti strutturali di HA secondo i dati della spettroscopia NMR, %
Torba HA s e □= C 1 1 0 1 Chp □= 1 0 e □= C 1 1 0 1 o< □= 1 о < о < 1 0 1 о Г си 1? 1 1? 1
Iniziale 25,0 5,3 8,1 3,0 21,4 19,8 4,8 3,6 3,0 6,0 0,275
Termotrattato 22,0 3,2 4,1 3,3 19,5 33,3 3,3 2,4 2,4 6,5 0,456
Nota: " - aromaticità
Dai cromatogrammi sono stati calcolati i tempi di ritenzione (T), s e i valori dei volumi di ritenzione corretti:
dove I è la distanza sul cromatogramma dal momento in cui il campione viene iniettato nella colonna al momento in cui esce il picco massimo, m; U - velocità del nastro grafico, m/s.
Tabella 4. Tempi di ritenzione dei composti organici su HA durante il riscaldamento lineare da 333 a 363 K
Adsorbati Peso molecolare Punto di ebollizione, °С Momento di dipolo Polarizzabilità, А3 Adsorbente HA Tempi di ritenzione, s
Pentano 72,2 36,1 0 10,0 rif. 16.7
Esano 86,2 68,7 0 11,9 rif. 21.9
Eptano 100,2 93,6 0 13,7 rif. 29.7
Isoottano 114,2 99,3 0 rif. 34.9
Cicloalcani
Cicloesano 84,2 81 0 11,0 rif. 28.1
Eptene 98,2 93,6 rif. 29.5
Eteri
Etere dietilico 74,1 35,6 1,18 10,0 rif. 18.5
Etere dipropilico 102,2 91,0 13,7 rif. 21.5
esteri
Acetato di etile 88,1 77,2 1,81 9,0 rif. 37.7
formiato di butile rif. 43.6
Eteri ciclici
Diossano 88,1 101,3 0 9,6 rif. 39.9
Acetone 58,1 56,2 1,66 6,6 rif. 21.1
Metiletilchetone 72,1 79,6 rif. 20.2
Butanolo-2 74,1 99,5 1,65 9,5 rif. 47.2
aromatico
Benzene 78,1 80,1 0 10,4 rif. 29.1
Toluene 92,1 110,6 0,36 12,4 rif. 34.2
Sostituito con cloro
Tetracloruro di carbonio 153,8 76,8 11,2 rif. 14.3
V \u003d Schyarglt P0t,
dove W1 è la velocità volumetrica del gas di trasporto, m/s; P1, T1 - pressione e temperatura nel flussometro del gas di trasporto, Pa e K; P0 - pressione del gas all'uscita della colonna, Pa; T - temperatura della colonna, K; ] - correzione della caduta di pressione nella colonna; t è il peso dell'adsorbente, kg.
] = 3[(P/P0)2 -1]/2[(P/P0)3-1], dove P1 è la pressione del gas all'ingresso della colonna, Pa.
Lo studio delle caratteristiche termodinamiche dell'adsorbimento si è basato sul soddisfacimento della seguente condizione: l'equilibrio gas-adsorbente deve essere stabilito in non più di 60 s. La condizione cromatografica di equilibrio per HA, come hanno dimostrato gli studi, corrisponde a picchi simmetrici. Questi autori hanno scoperto che la velocità del gas di trasporto e la dimensione del campione di adsorbato non avevano alcun effetto sui volumi di ritenzione, ad es. L'equilibrio termodinamico è raggiunto nel sistema.
I volumi di ritenzione calcolati a diverse temperature hanno permesso di calcolare i calori di adsorbimento e altre caratteristiche termodinamiche in condizioni di equilibrio.
La base del metodo gascromatografico è l'idea di stabilire un equilibrio gas - fase condensata per un adsorbato caratterizzato da un coefficiente di distribuzione K:
Il calore di adsorbimento (entalpia) è stato determinato dalla formula:
AN \u003d R e 1n (Kd / T), kJ / mol.
L'entropia di adsorbimento è stata determinata dall'equazione А5=(АА-АО)/T, J/molK, dove AO è l'energia libera di adsorbimento (energia di Gibbs) -АО=JT 1pK, kJ/mol.
risultati e discussione
Dal punto di vista della teoria statistico-molecolare dell'adsorbimento di HA dovuto alla presenza di gruppi carbossilici, idrossili fenolici, chinoidi, gruppi carbonilici, chetoni, gruppi carbonilici, aldeidi ed altri, a quanto pare, può essere attribuito a un assorbente. Allo stato solido, le molecole planari di HA sono "impacchettate" in pacchi di più strati, che è una manifestazione locale di ordinamento parziale. Il sistema di policoniugazione, dovuto alla delocalizzazione degli elettroni x, porta ad un aumento dell'influenza reciproca degli atomi, ma la presenza di gruppi diversi crea ancora una disomogeneità chimica della superficie, che è associata a una debole specificità.
Come si evince dai dati riportati in tabella. 4, i tempi di ritenzione per quasi tutti gli adsorbati su HA dalla torba trattata termicamente sono più brevi di quelli su HA dalla torba originale.
I maggiori volumi trattenuti si osservano negli alcoli, ciclici ed esteri, aromatici; il più piccolo - in alcani, cloro-sostituiti, chetoni ed eteri.
La teoria statistica molecolare dell'adsorbimento mette in relazione i tempi di ritenzione ei volumi di ritenzione con le interazioni elettrostatiche intermolecolari dei dipoli. Dunque,
L'immagine nativa per diverse classi di composti organici è determinata dalla presenza o assenza di momenti di dipolo nelle molecole. Come è noto, le molecole di alcoli, esteri e aromatici hanno un momento di dipolo significativo e gli alcani hanno un momento di dipolo zero. Tuttavia, è impossibile mettere in relazione inequivocabilmente i tempi di ritenzione con il momento di dipolo delle sostanze. Ad esempio, l'acetone ha un momento di dipolo di 1,66 e il toluene di 0,36, mentre il tempo di ritenzione dell'acetone è molto più breve di quello del toluene.
Probabilmente, in questo caso, non solo le interazioni intermolecolari, ma anche elettrostatiche svolgono un ruolo nell'interazione di adsorbimento, ma un grande contributo è dato dall'interazione aspecifica dell'adsorbente con l'adsorbato, che è determinata dai valori del van der Waals raggi e i valori della polarizzabilità, che per il toluene (Tabella 4) è quasi 2 volte superiore rispetto all'acetone. Ciò è spiegato dalla struttura porosa disomogenea di HA. Gli studi hanno dimostrato che il raggio dei pori di HA varia entro 10,70 Å con la predominanza di piccoli pori di 10,15 Å, che è commisurato alle dimensioni lineari del frammento "primario" della struttura HA. Per il toluene, il diametro della molecola è molto più piccolo, quindi le sue molecole penetrano facilmente nei pori dell'adsorbente.
Dal tavolo. 4 si può vedere che non vi è alcuna variazione regolare dei valori dei volumi trattenuti dal punto di ebollizione dei composti organici. Ciò è spiegato dal fatto che il punto di ebollizione è associato all'interazione delle molecole tra loro nel liquido e, nel caso dell'adsorbimento, l'interazione avviene con l'adsorbente.
A giudicare dai dati ottenuti, gli alcani mostrano, in media, una bassa capacità di adsorbimento, che è notevolmente superiore per gli HA della torba trattata termicamente. Tra gli alcani, l'isoottano ha volumi di ritenzione leggermente superiori. Gli alcani che hanno legami st interagiscono in modo non specifico con gli adsorbenti. Aumentano linearmente i valori delle polarizzabilità elettroniche nella serie degli alcani dal pentano all'esano e aumentano anche i volumi di ritenzione (RV).
La ciclizzazione della catena alcanica porta ad una diminuzione dei volumi di cicloesano a causa di una diminuzione del numero di atomi di idrogeno e di una deviazione nella disposizione degli atomi di carbonio dalla complanarità. I collegamenti dello scheletro di carbonio probabilmente non possono toccare contemporaneamente la faccia basale dell'adsorbente.
Si osservano SV molto elevati per gli idrocarburi aromatici, il toluene in misura maggiore. Inoltre, i valori sono ugualmente alti per entrambi i tipi di HA. Questo comportamento del toluene può essere spiegato dalla presenza di un gruppo metilico, che, a causa della manifestazione di un effetto elettronico induttivo positivo e dell'effetto di sovraconiugazione, aumenta la densità elettronica nell'anello benzenico e la riduce sul gruppo metilico.
Gli alcoli con un grande momento di dipolo hanno valori di SV elevati, che aumentano soprattutto durante l'adsorbimento su HA torbato trattato termicamente.
Chetoni ed eteri, in quanto sostanze con polarità più debole, hanno SV più basso. Ciò è dovuto al minor contributo dell'energia del legame idrogeno alla ritenzione di chetoni ed eteri, sebbene il momento di dipolo, ad esempio, dell'acetone sia uguale al momento di dipolo dell'alcol butilico.
Gli esteri ciclici sono caratterizzati dal più alto SV, a causa di una polarizzazione più pronunciata dei legami nei frammenti contenenti ossigeno rispetto agli eteri e, di conseguenza, una maggiore capacità di formare legami idrogeno.
Tuttavia, in tutti questi casi, viene preservata l'identità chimica della molecola, cioè l'interazione ha un carattere "molecolare" piuttosto che "chimico".
Come notato sopra, le proprietà di adsorbimento dell'HA della torba trattata termicamente sono superiori a quelle dell'HA originale, che è più pronunciato nel caso degli adsorbati polari. Questa natura delle proprietà può essere completamente spiegata dai cambiamenti che si verificano con HA nel processo di termolisi a bassa temperatura della torba. Secondo i dati delle analisi chimiche e della spettroscopia NMR, si osserva un leggero aumento dei gruppi contenenti ossigeno (idrossili carbossilici, fenolici) e dei frammenti di glucosio.
Come si evince dai dati in Tabella. 5, i calori di adsorbimento per molecole dipoli (eteri, chetoni) e per molecole debolmente dipoli (idrocarburi aromatici e alcoli) sono superiori ai calori di adsorbimento di n-alcani, che hanno momento di dipolo zero e sono incapaci di una specifica molecola interazione. Va notato, come hanno sottolineato gli autori, che il calore totale di adsorbimento di qualsiasi molecola organica è costituito da due componenti: il calore di adsorbimento dovuto all'interazione con i centri attivi dell'adsorbente e il calore di interazione delle molecole adsorbite con ciascuna Altro. Tuttavia, non è possibile separare e calcolare le batterie da questi risultati.
Dai dati sperimentali si evince che, nelle serie degli n-alcani, un aumento della lunghezza della catena di carbonio porta ad un aumento del calore di adsorbimento e della loro polarizzabilità. Il valore del calore di adsorbimento per n-al-kanes è commisurato ai valori dell'energia di interazione di van der Waals (<5 кДж/моль), вероятно, взаимодействие между ГК и н-алканами осуществляется за счет ван-дер-ваальсовых сил.
Dai dati in tabella. La Figura 5 mostra che i calori di adsorbimento di eteri, alcoli, chetoni e composti aromatici su HA si trovano entro 5 kJ/mol, che sono tipici per le energie dei tipici legami idrogeno; pertanto, l'adsorbimento procede attraverso la formazione di legami idrogeno.
Tabella 5. Caratteristiche termodinamiche dei volumi di adsorbimento e ritenzione
Adsorbati Adsorbenti, HA Volume di ritenzione a 333,363 K, ^■103, m3/kg -AN, kJ/mol -A5, J/mol -AG, kJ/mol
Pentano rif. 4.8 1.9 10.1 5.3
ter. 9.3 3.8 19.5 10.2
Esano rif. 6.2 2.5 13.0 6.8
ter. 11.2 4.5 23.5 12.2
Eptano rif. 9,0 3,6 18,9 9,9
ter. 13.2 5.3 27.7 14.5
Isoottano rif. 11.5 4.6 24.1 12.6
ter. 16,7 6,7 35,0 18,3
Cicloalcani
Cicloesano rif. 2.3 1.0 4.8 2.5
ter. 9.3 3.8 19.5 10.2
Eptene rif. 8.4 3.4 17.6 9.2
ter. 10.1 4.1 21.2 11.1
Eteri
Etere dietilico rif. 6.8 2.7 14.3 7.5
ter. 13,5 5,4 28,3 14,8
Etere dipropilico rif. 11.5 4.6 24.1 12.6
ter. 17,4 7,0 36,5 19,1
esteri
Acetato di etile rif. 19,7 8,0 41,3 21,6
ter. 28,2 11,4 59,1 30,9
formiato di butile rif. 24,3 9,8 51,0 26,7
ter. 30,5 12,3 64,0 33,5
Eteri ciclici
Diossano rif. 26,5 10,7 55,6 29,1
ter. 27,8 11,2 58,3 30,5
Acetone rif. 10.1 4.1 21.2 11.1
ter. 14,3 5,8 30,0 15,7
Metiletilchetone rif. 9.7 3.9 20.3 10.6
ter. 10.1 4.0 21.1 11.0
Butanolo-2 rif. 39,2 15,8 82,2 43,0
ter. 40,2 16,2 84,3 44,1
aromatico
Benzene rif. 18.4 7.4 38.6 20.2
ter. 19.2 7.7 40.3 21.1
Toluene rif. 20.2 8.1 42 .4 22.2
ter. 25,4 10,2 53,3 27,9
Sostituito con cloro
Tetracloruro di carbonio rif. 4.2 1.7 8.8 4.6
ter. 8.4 3 , 4 17.6 9.2
L'etere dietilico è caratterizzato da un basso calore di adsorbimento paragonabile a quello dell'esano. Probabilmente, la manifestazione di una forte interazione specifica dei gruppi funzionali di HA con etere dietilico è ostacolata dalla localizzazione in
ossigeno nel mezzo della catena degli idrocarburi, che rende difficile il contatto con l'adsorbente. Per le molecole di estere, i calori di adsorbimento sono maggiori rispetto agli eteri a causa della presenza di gruppi C=0, che conferiscono una maggiore polarità, e vi è un contatto più stretto con i gruppi funzionali dell'adsorbente. Sulla superficie degli HA, probabilmente, la densità elettronica è localmente concentrata alla periferia dei gruppi funzionali, il che garantisce l'elevata specificità dell'adsorbimento di molecole di alcoli, eteri complessi e ciclici e composti aromatici. Come notano gli autori, è necessario tenere conto dell'effetto del legame idrogeno sul calore di adsorbimento dell'adsorbato-adsorbente. Il calore di adsorbimento di sostanze che formano legami a idrogeno sarà sempre maggiore del calore di adsorbimento di sostanze di struttura simile, ma non lo formano. Ad esempio, l'etere dipropilico ha un calore di adsorbimento maggiore rispetto all'etere dietilico a causa di un legame idrogeno più forte. Le molecole di HA agiscono come donatori di protoni (accettore di elettroni a causa dei gruppi OH- e, in misura minore, COOH) e le molecole di etere ed estere agiscono come donatori di elettroni (accettore di protoni), a causa di un legame etereo (-O-) con la formazione di un associato, ma in questo caso non si verifica la transizione completa del protone. Le proprietà del donatore di elettroni del legame etereo dell'etere dipropilico sono superiori a quelle dell'etere dietilico. Pertanto, il contributo al calore di adsorbimento dovuto al legame idrogeno è maggiore per l'etere dipropilico. Va notato che gli HA della torba trattati termicamente sono probabilmente caratterizzati da una maggiore densità elettronica alla periferia dei gruppi funzionali e proprietà di ritiro degli elettroni rispetto agli HA della torba originale.
È noto che il calcolo dell'entropia di adsorbimento viene effettuato per stabilire il grado di mobilità delle molecole adsorbite. Il cambiamento di entropia include l'entropia del movimento traslazionale, rotatorio e vibrazionale delle molecole.
Secondo i dati (Tabella 5), esiste una relazione tra |-AN| e |-A6| per diverse sostanze: idrocarburi alifatici, aromatici, alcoli, eteri e chetoni. Si può presumere che l'interazione degli adsorbati elencati con HA abbia la stessa immagine. Grandi valori negativi sono tipici per alcoli, eteri complessi e ciclici, che è associato alla pronunciata polarità delle molecole. Per la torba HA trattata termicamente, la caratteristica di entropia negativa è inferiore rispetto alla torba originale. Probabilmente, nella struttura della torba HA trattata termicamente c'è una distribuzione più ampia in termini di spostamento, rotazione e vibrazione delle molecole di adsorbato. Secondo i dati, più il volume molare dell'adsorbato è vicino al volume limite dello spazio di assorbimento dell'adsorbente, più inibito è il movimento di traslazione e rotazione della molecola di adsorbato, maggiore è il valore assoluto dei valori negativi di Ae
Per gli acidi umici, il valore del volume di assorbimento è 4,0,10-4 m3/kg, che è vicino ai volumi molari di butanolo-2, acetato di etile, diossano, benzene e toluene, compresi tra 2,5 e 3,0,10-4 m3/kg, quindi, sono caratterizzati da bassi valori di A£ Per n-alcani, alcheni e idrocarburi cloro-sostituiti i volumi molari sono inferiori a 2.5.10-4 m3/kg, per i quali i valori di A£ sono più alto.
I valori dell'energia di Gibbs indicano la possibilità del processo di adsorbimento, nonché lo stato di equilibrio del sistema. I valori di AO più elevati sono stati ottenuti per alcol, esteri ciclici e idrocarburi aromatici. Se confrontiamo i valori AO per gli HA della torba originale e trattata termicamente, quest'ultimo valore è leggermente più alto. È probabile che il processo di adsorbimento sugli HA della torba originale sia più spostato verso il desorbimento rispetto alla torba trattata termicamente.
Un'analisi delle caratteristiche termodinamiche dell'adsorbimento suggerisce che gli adsorbati possono essere disposti in fila al diminuire della loro capacità di adsorbimento: alcoli > esteri > eteri ciclici > chetoni aromatici > eteri, alcheni, alcani
1. È stato dimostrato che i centri di adsorbimento attivi nell'HA sono gruppi funzionali: idrossili carbossilici, fenolici, frammenti glucosidici e aromatici. Poiché gli HA della torba trattata termicamente hanno un alto contenuto dei gruppi di cui sopra, hanno una maggiore capacità di adsorbimento.
2. È stato dimostrato che la capacità di adsorbimento dell'HA rispetto ai composti polari (alcoli, eteri complessi e ciclici, aromatici, chetoni) è superiore a quella degli adsorbati non polari (alcani, alcheni).
3. Si ottengono le relazioni tra alcune caratteristiche fisico-chimiche (polarizzabilità, momento di dipolo) degli adsorbati e parametri di ritenzione.
4. È stato dimostrato che la maggiore capacità di adsorbimento dell'HA di torba trattata termicamente è spiegata dall'aumento del contenuto di gruppi contenenti ossigeno (idrossili carbossilici, fenolici), glucoside e frammenti aromatici nella struttura rispetto all'HA originale.
5. È stato rivelato che le caratteristiche termodinamiche (-AD-A^Ab) per gli HA della torba originale e trattata termicamente sono interconnesse per tutti gli adsorbati studiati.
6. È stato stabilito che la probabilità termodinamica di adsorbimento su HA avviene nel seguente ordine: alcoli > esteri > eteri ciclici > chetoni aromatici > eteri, alcheni, alcani.
BIBLIOGRAFIA
1. Komissarov ID, Loginov L.F. Sostanze umiche nella biosfera. - M.: Nauka, 1993. - 352 pag.
2. Lishtvan I.I., Kruglitsky N.N., Tretinnik V.Yu. Meccanica fisico-chimica delle sostanze umiche. - Minsk: Scienza e tecnologia, 1976. - 264 p.
3. Pal Regno Unito, Chakravarti SK Assorbimento in volume del complesso etildiamminico Co sul suolo e acidi umici di torba // Journal of Indian Chemical Society. - 1986. - V. 63. - N. 10. - P. 883-889.
4. Pilipenko A.T., Vasiliev N.G., Buntova M.A., Savkin A.G. Meccanismo e forza di assorbimento dei cationi di metalli di transizione da parte degli acidi umici // Doklady AN UkrSSR. - 1986. - N. 7. - S. 42-45.
5. Gamayunov N.I., Maslennikov BI, Shulman Yu.A. Scambio ionico negli acidi umici // Chimica dei combustibili solidi. -1991. - N. 3. - S. 32-39.
6. Alexandrov IV, Kandelaki GI, Kulikova I.P. Assorbenti zeolite-umici per il trattamento delle acque reflue // Chimica dei combustibili solidi. - 1994. - N. 4-5. - S. 136-142.
7. Bratasevszskij A., Gaidarob O., Gordienko Sz. Studio del processo di formazione complessa di acidi umici con il metodo potenziometrico // Agrochem. es tobaj. - 1971. - V. 2. - N. 2.
8. Parkhomenko V.V., Kudra A.A. Sul calcolo delle funzioni termodinamiche del processo di adsorbimento di alcol metilico da parte di acidi umici e umati secondo un'isoterma // Fenomeni superficiali in sistemi dispersi. - Kiev: Naukova Dumka, 1974.
Problema. 3. - S. 35-43.
9. Tarnovskaya L.I., Maslov S.G., Smolyaninov S.I. Composizione chimica delle sostanze organiche dei residui solidi della pirolisi della torba // Chimica dei combustibili solidi. - 1988. - N. 3. - S. 26-29.
10. Lishtvan I.I., Korol N.T. Proprietà di base della torba e metodi per la loro determinazione. - Minsk: Scienza e tecnologia, 1975. - 320 p.
11. Bazin E.T., Kopenkin V.D., Kosov V.I. ecc. Analisi tecnica della torba. - M.: Nedra, 1992. - 431 pag.
12. Tarnovskaya LI, Maslov S.G. Cambiamenti nella composizione chimica degli acidi umici durante la termolisi della torba // Chimica dei combustibili solidi. - 1994. - N. 4-5. - S. 33-39.
13. Kiselev AV, Yashin Ya.I. Applicazioni fisiche e chimiche della gascromatografia. - M.: Chimica, 1973. - 214 p.
14. Wigdergauz MS, Izmailov RI Applicazione della gascromatografia per determinare le proprietà fisico-chimiche delle sostanze.
M.: Nauka, 1970. - 159 pag.
15. Stallo D., Wesram E., Zinke G. Termodinamica chimica dei composti organici. - M.: Mir, 1971. - 807 pag.
AUMENTARE L'EFFICIENZA DELL'OPERAZIONE INDUSTRIALE DELL'UNITÀ DI REFORMING LCH-35-11/1000 E LG-35-8/300B IN KINEF SULLA BASE DI UN SISTEMA DI CONTROLLO DEL FUNZIONAMENTO DEL CATALIZZATORE
DI. Melnik, SA Galushin, AV Kravtsov, E.D. Ivanchina, V.N. Fetisova
E-mail del Politecnico di Tomsk: [email protetta]
Viene presa in considerazione la prospettiva di utilizzare il sistema di controllo del processo automatizzato del sistema di controllo del funzionamento del catalizzatore basato su reti e banche dati di informazioni di fabbrica. Viene mostrata la possibilità di ridurre la formazione di coke quando si lavora a un'attività ottimale utilizzando il metodo della modellazione matematica. Viene descritto lo schema esistente e sviluppato per automatizzare l'acquisizione e l'analisi dei dati tecnologici necessari per i calcoli.
L'efficienza della produzione industriale dipende in maniera determinante dalla controllabilità dei processi tecnologici, in primis, dalla possibilità di accesso tempestivo agli indicatori di prestazione del catalizzatore e dalla fornitura di controllo, analisi e previsione dei parametri tecnologici del processo.
Le reti informative di fabbrica di un sistema automatizzato di controllo dei processi (APCS) risolvono solo i problemi di raccolta, archiviazione, accumulazione, strutturazione dei dati con la successiva fornitura di queste informazioni a quegli utenti le cui decisioni dovrebbero basarsi su di esse. APCS combina un gran numero di sistemi distribuiti in un unico spazio informativo. Il livello inferiore di questo sistema è rappresentato dai server di comunicazione che svolgono le funzioni di controllo separatore
reti di comunicazione e informazione e il trasferimento di informazioni tecnologiche al livello successivo. Nella zona della rete informativa, che copre l'intera azienda, è presente un server per la raccolta delle informazioni tecnologiche, che consente di memorizzare grandi quantità di dati sul processo tecnologico. Gli utenti hanno accesso sia alle informazioni archiviate sul server che alle informazioni in tempo reale sui server di comunicazione. Al fine di riassumere le informazioni provenienti da varie fonti, LLC PO Kirishinefteorgsintez ha sviluppato e implementato, insieme alla società di ingegneria specializzata Sevzapmontazhavtomatika, un pacchetto software - l'Unified Thematic Data Mart (ETVD), che fornisce all'utente una comoda interfaccia grafica per l'accesso dati di controllo di laboratorio, caratterizzandoli e la loro rappresentazione combinata.
Definizioni di base e metodi per classificare i processi di adsorbimento.
L'adsorbimento si riferisce a fenomeni che si verificano a seguito di una diminuzione spontanea dell'energia superficiale.
Adsorbimento– il processo di ridistribuzione spontanea reversibile o irreversibile dei componenti di un sistema eterogeneo tra lo strato superficiale e il grosso della fase omogenea.
Nei sistemi multicomponente, allo strato superficiale è preferibile il componente che abbassa la tensione interfacciale. Nei sistemi monocomponenti, durante la formazione dello strato superficiale, la sua struttura cambia (un certo orientamento di atomi e molecole, polarizzazione), chiamata autoassorbimento.
Viene chiamata la fase più densa su cui sono localizzate le interazioni di adsorbimento assorbente. La sostanza ridistribuita tra il volume della fase omogenea e lo strato superficiale è indicata con il termine " adsorbito».
In alcuni casi, il processo di adsorbimento è reversibile. In questo caso, in determinate condizioni, parte delle molecole adsorbite può passare dallo strato superficiale nel volume della fase per effetto di fenomeni di cinetica molecolare. Viene chiamato il processo inverso di adsorbimento desorbimento.
Metodi per classificare i processi di adsorbimento.
Classificazione dei processi di adsorbimento in base allo stato di aggregazione delle fasi interagenti. A seconda dello stato di aggregazione delle fasi adiacenti si distinguono le seguenti tipologie di processi di adsorbimento:
Adsorbimento di gas su adsorbenti solidi;
Adsorbimento di sostanze disciolte all'interfaccia "solido-liquido" e "liquido-liquido";
Adsorbimento di tensioattivi all'interfaccia liquido-gas.
Classificazione dei processi di adsorbimento in base al meccanismo di interazione dell'adsorbente e dell'adsorbato. L'adsorbimento può essere considerato come l'interazione delle molecole di adsorbato con i centri attivi dell'adsorbente. In base al meccanismo della loro interazione, si suddividono i seguenti tipi di adsorbimento:
1) adsorbimento fisico (molecolare).- l'interazione tra le molecole dell'adsorbato e dell'adsorbente avviene per effetto delle forze di van der Waals, legami idrogeno (senza il verificarsi di reazioni chimiche);
2) adsorbimento chimico (chemisorbimento)– l'adesione di molecole di adsorbato ai centri attivi dell'adsorbente avviene a seguito di reazioni chimiche di vario tipo (ad eccezione delle reazioni di scambio ionico);
3) adsorbimento a scambio ionico (scambio ionico) - la ridistribuzione della sostanza adsorbita tra la soluzione e la fase solida (scambiatore ionico) secondo il meccanismo delle reazioni di scambio ionico.
Per una descrizione quantitativa dei processi di adsorbimento vengono utilizzate due grandezze.
1) Assorbimento assolutoè la quantità (mol) o la massa (kg) dell'adsorbato per unità di superficie o massa dell'adsorbente. Designazione - A; dimensione: mol / m 2, mol / kg, kg / m 2, kg / kg.
2) Adsorbimento di Gibbs (eccesso).è l'eccesso di sostanza adsorbata nello strato superficiale di un certo spessore rispetto alla sua quantità nel volume della fase omogenea, per unità di superficie o massa dell'adsorbente. Designazione - G; unità: mol/m 2 , mol/kg.
La relazione tra adsorbimento assoluto ed eccesso può essere illustrata utilizzando l'equazione:
G \u003d A - c * h (3.1)
dove c è la concentrazione di equilibrio della sostanza nel volume della fase, mol/m3;
h è lo spessore dello strato superficiale, preso condizionatamente pari a 10 -9 m.
Nei sistemi eterogenei multicomponenti, quando l'uno o l'altro componente viene ridistribuito tra il volume di una fase omogenea e lo strato superficiale, vale l'equazione per l'energia interna in eccesso della superficie:
U = T * S + s * s + Sm io * n io (3.2)
Portando tutti i termini dell'equazione nell'area unitaria della superficie interfacciale, otteniamo:
U s = T * S s + s + Sm i * Ã i (3.3)
dove à i = n i / s è l'eccesso della i-esima componente nello strato superficiale, cioè l'adsorbimento di Gibbs.
Per un sistema a una componente, l'equazione (3.3) assume la forma:
G s = s + m * Ã (3.4)
dove G s = U s - T * S s è l'energia di Gibbs della superficie o il lavoro di creazione di un'area unitaria della superficie;
m * Г - compattazione della sostanza della sostanza adsorbita nello strato superficiale.
Sulla base dell'equazione (3.4), possiamo concludere che durante l'adsorbimento, il lavoro per creare una superficie interfacciale consiste nel lavoro di formare una superficie (rompendo i legami coesivi nella maggior parte della fase di adsorbato) e nel compattare la sostanza nello strato superficiale.
Nello stato di equilibrio dinamico tra l'adsorbente e l'adsorbato, la variazione dell'energia di Gibbs di un sistema eterogeneo ΔG = 0, la termodinamica del processo di adsorbimento è descritta da un'equazione chiamata Equazione fondamentale di adsorbimento di Gibbs:
Ds = SÓ i * dm i (3.5)
Questa equazione è universale, poiché è valida per tutti i tipi di processi di adsorbimento
Casi particolari dell'equazione di adsorbimento di Gibbs.
1) Adsorbimento da soluzioni.
Per il potenziale chimico dell'i-esimo componente del sistema durante l'adsorbimento alle interfacce "liquido - solido adsorbente" e "liquido - gas" valgono le seguenti equazioni:
m io = m io 0 + R*T*ln un io (3.6)
dm i = R*T* d ln a i (3.7)
dove m i 0 è il potenziale chimico dell'i-esimo componente del sistema in condizioni standard;
a i – attività della i-esima componente del sistema in condizioni standard.
Sulla base di ciò, l'equazione di adsorbimento di Gibbs assumerà la forma:
à i = - un i / R*T * (ds / da i) (3.8)
Per soluzioni non elettrolitiche, prendiamo a i \u003d c i, quindi:
à i \u003d - s / R * T * (ds / ds) (3.9)
Per soluzioni elettrolitiche:
à i = - ñ ± n / R*T * (ds / dñ ± n) (3.10)
dove c ± è la concentrazione ionica media della soluzione;
n è il coefficiente stechiometrico.
2) Adsorbimento di sostanze dalla fase gassosa.
Secondo l'equazione di Mendeleev-Claiperon:
P \u003d c * R * T (3.11)
A questo proposito, l'equazione di Gibbs per l'adsorbimento dei gas sugli adsorbenti solidi è scritta nella forma seguente:
à i = - Р / R*T * (ds / dР) (3.12)
In pratica, l'equazione di adsorbimento di Gibbs permette di calcolare la quantità di adsorbimento di sostanze nello strato interfacciale, per il quale viene determinata la tensione superficiale, in base alla misura della tensione superficiale a vari valori di concentrazione del liquido o pressione di equilibrio del gas .


